A Microdeletion at Xp22.13 Associated with Nance-Horan Syndrome Identified by Copy Number Variation Sequencing
Donglan Sun1#, Qing Guo1#, Wenqi Chen1, Jing Zhang1, Yuanyuan Peng1, Ke Wang1, Jing Zhang1*
1Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital, Shijiazhuang, Hebei Province, China
#Both authors contributed equally to this manuscript.
*Correspondence to: Jing Zhang, PhD, Professor, Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital, Key Laboratory of Maternal and Fetal Medicine of Hebei Province, 16 Tangu-North Street, Shijiazhuang 050011, Hebei Province, China; E-mail: zhangjing_hbyd_81@126.com
Abstract
Nance-Horan syndrome (NHS) represents the uncommon X-linked genetic disease with the features of congenital cataracts, tooth deformities, mental retardation along with facial deformities. It is mostly caused by small insertions, deletions, and nonsense mutations of NHS gene on Xp22.13, which may cause the NHS protein truncation. The present work discovered the 1.62-Mb microdeletion on Xp22.13 through copy number variation sequencing in a China girl with NHS syndrome, which included these genes: NHS, Cyclin-dependent kinase-like 5, Scm polycomb group protein like 2, Retinoschisin1, Scm polycomb group protein like 1, RALBP1 associated Eps domain containing 2, and Retinoic acid induced 2. Her parents did not find the microdeletion of Xp22.13, which was a novel microdeletions of Xp22.13 will cause haploinsufficiency of flanking genes, non-random X chromosome inactivation may increase the X-linked disease risk.
Keywords: Nance-Horan syndrome, copy number variation sequencing, Xp22.13 microdeletion, mental retardation, congenital cataract
1 INTRODUCTION
Nance-Horan syndrome (NHS) [MIM 202350], with the features of congenital cataracts in both eyes, dysmorphic facial features, dental abnormalities like bud molars or screwdriver-shaped teeth, with occasional brachymetacarpia and mental retardation, represents the uncommon X-linked recessive hereditary disorder initially put forward in 1974 by Nance et al[1]. NHS gene is mapped onto Xp22.13 at the interval of 1.3-Mb[2]. Its gene includes the genomic DNA (gNDA) of 650kb, which encodes one candidate nuclear protein of 1,630 amino acids. Research discovers a complex expression pattern of temporal and spatial regulation, accompanied by pleiotropic characteristics in NHS, indicating the role of the gene in regulating eye, teeth, craniofacial and cerebral development[3]. NHS can be inherited on the X chromosome, and female heterozygous carriers typically show a close yet moderate phenotype to infected men[4,5], usually with posterior Y-suture lens opacities and innate cortical riders, with no congenital cataract[6].
The present work clinically and genetically analyzed the 1.62Mb aninterstitial microdeletion on Xp22.13 detected by next generation sequencing in a 7 years old girl, who showed severe mental retardation, cataract, abnormal tooth development, low muscle tone, and severe epilepsy. The parents were not found to have chromosome copy number variation (CNV), as a result, X-linked deletion was derived from her daughter de novo. Furthermore, the girl’s cytogenetic analysis revealed an apparently karyotype of 46, XX, t(17; 18)(q23; p11.3) that was transmitted from the carrier mother
2 CASE PRESENTATION
2.1 Clinical Report
A 7 years old girl. It is the third birth of her mother, full term delivery, birth weight 3.1kg, no asphyxia. The fundus screening after birth diagnosed congenital cataract in her right eye (Figure 1). When she was 5 months the epilepsy began. After the antiepileptic treatment, the frequency of seizures and clinical symptoms have not been significantly improved (3-4times/day, 2-3min/time). The child's psychomotor development is seriously backward. She can't lift her head, turn over, sit, stand, walk or speak. Her hands can’t grasp. She neither knows her family nor have eye contact. Physical examination: body length 120cm, weight 10.5kg (<3SD), underdeveloped, poor nutrition, flat occipital fovea, congenital cataract in the right eye, large teeth gap, cardiopulmonary (-), low muscle tension of limbs, weak tendon reflex. Auxiliary examination: no obvious abnormality was found in chest X-ray, abdominal ultrasound, cardiac ultrasound and head magnetic resonance imaging. The parents were healthy, married without close relatives, denied the history of infectious diseases during pregnancy, denied the history of radiation exposure and family genetic history. With the approval of hospital medical ethics committee (No. 20220165) and the informed consent of the parents of the children, the peripheral blood samples of the children and their parents were collected for G-banding karyotype analysis, CNV sequencing (CNVseq) and fluorescence quantitative polymerase chain reaction (PCR) analysis.
|

Figure 1. Image of fundus screening of the patient.
3 MATERIALS AND METHODS
3.1 Chromosome Analysis
By adopting standard cytogenetic approaches, this work cultivated peripheral blood in serum-free lymphocyte medium for a 72-h period. Later, G-banding was adopted in chromosome analysis with trypsinization as well as Giemsa’s staining. This work utilized altogether 20 metaphase spreads for analysis[7], and depicted karyotypes in line with the international system for human cytogenomic nomenclature (ISCN 2016).
3.2 Detection of Chromosomal CNVseq by Low Coverage Whole Genome High Throughput Sequencing
This work conducted CNVseq for detecting chromosome anomaly by low-coverage whole genome sequencing. In brief, gDNA was confirmed through 2 approaches together: (1) 1% agarose gels were utilized in DNA decomposition as well as contamination; (2) Qubit DNA assay kit was utilized to determine DNA contents with Qubit 2.0 Flurometer (Life Technologies, CA, USA). Thereafter, CLEANNGS DNA kit was adopted to produce sequencing library in line with specific protocols, followed by addition of index codes into every specimen. Later, by adopting Novaseq 5000/6000 S4 Reagent Kit (Illumina), those index-coded specimens were clustered onto the cBot cluster generation system in line with specific protocols. Following cluster production, Illumina NovaSeq 6000 platform was employed to sequence DNA libraries to generate the 150bp paired-end reads. Through primary quality control, this work first processed fastq-format raw reads. Then, BWA software was employed to compare clean reads against reference human genome (UCSC hg19), following by conversion of results to the bam format as well as sorting with samtools software. At last, basic information was statistically analyzed and compared. Thereafter, SNP/InDels were identified with Verita TreKKer. CNVs were detected by BIC-Seq and CNVnator, whereas SVs were discovered by Manta. Enliven was performed to do annotation for SNP/InDels/CNV/SV.
gNDA extraction Kit produced by Qiagen company in Germany was utilized to extract gDNA in line with kit protocols. DNA concentration was controlled at 50-250ng/μL. Then, sequencing libraries constructed were sequenced on the next-Seq CN500 platform. The sequencing results were analyzed, and each sequencing read was matched to its chromosome. Then the corresponding standardized Z-value analysis was done, and the chromosome abnormality was determined by Z-value. The clinical significance of CNV was determined by searching decipher, OMIM and DGV databases.
3.3 Fluorescence Quantitative PCR
Fluorescence quantitative PCR detection according to the results of high-throughput sequencing, the deletion region of the fragment was selected for fluorescent quantitative PCR verification. ABI7500 fluorescent quantitative PCR was used for relative quantitative analysis.
4 RESULTS
The cytogenetic analysis revealed an apparently karyotype of 46, XX, t (17;18) (q23;p11.3), which was inherited from her mother (Figure 2).
|
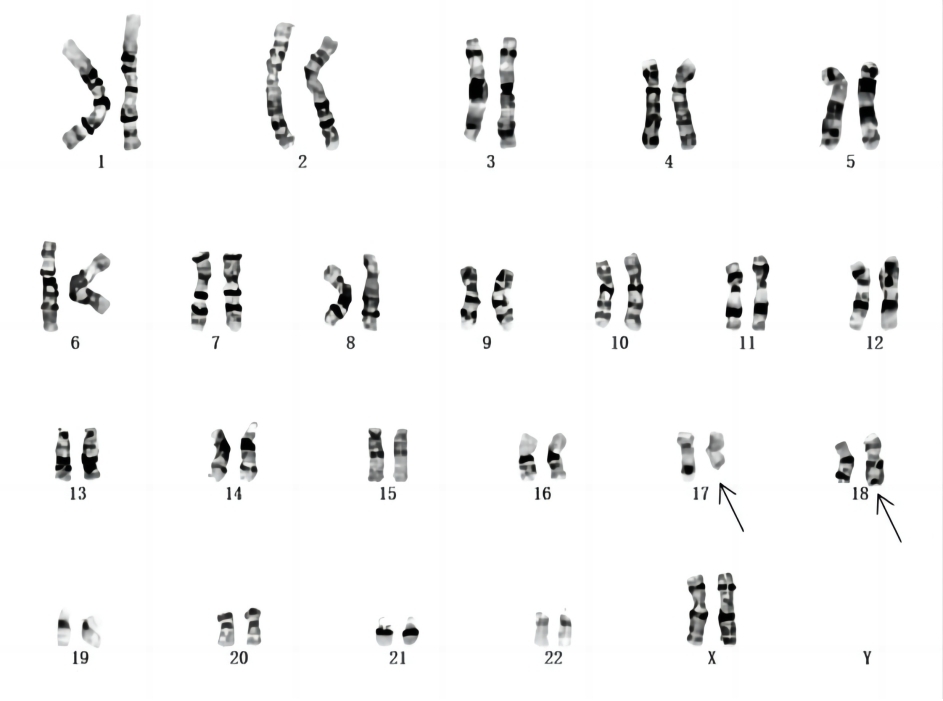
Figure 2. One balanced translocation that involved chromosomes 17 and 18 was identified by chromosome G-banding analysis within the child and her mother.
We found that there were 2 chromosomal copy variant loci were detected by CNV-seq (Figure 3) with the first deletion being at nucleotide positions 17160001-18780000 (GRCh37/hg19 assembly), which were matched with cytogenetic band Xp22.13. Its deletion length was predicted to be 1.62Mb, which involved these genes: NHS [OMIM:300457], Cyclin-dependent kinase-like 5 (CDKL5) [OMIM:300203], SCML2 [OMIM:300208], Retinoschisin1 (RS1) [OMIM:300839], SCML1 [OMIM:30027], RALBP1 associated Eps domain containing 2 (REPS2) [OMIM:300317], Retinoic acid induced 2 (RAI2) [OMIM:300217]. The second one was on nucleotide positions 22540001-22960000l (GRCh37/hg19 assembly), which were matched with cytogenetic band 14q11.2. Its deletion length was predicted to be 0.42Mb, referring to relevant literature and databases, there was no gene in this region and it was polymorphic. Her parents did not find the microdeletion of Xp22.13. The chromosome 14 polymorphism was inherited from his father.
|
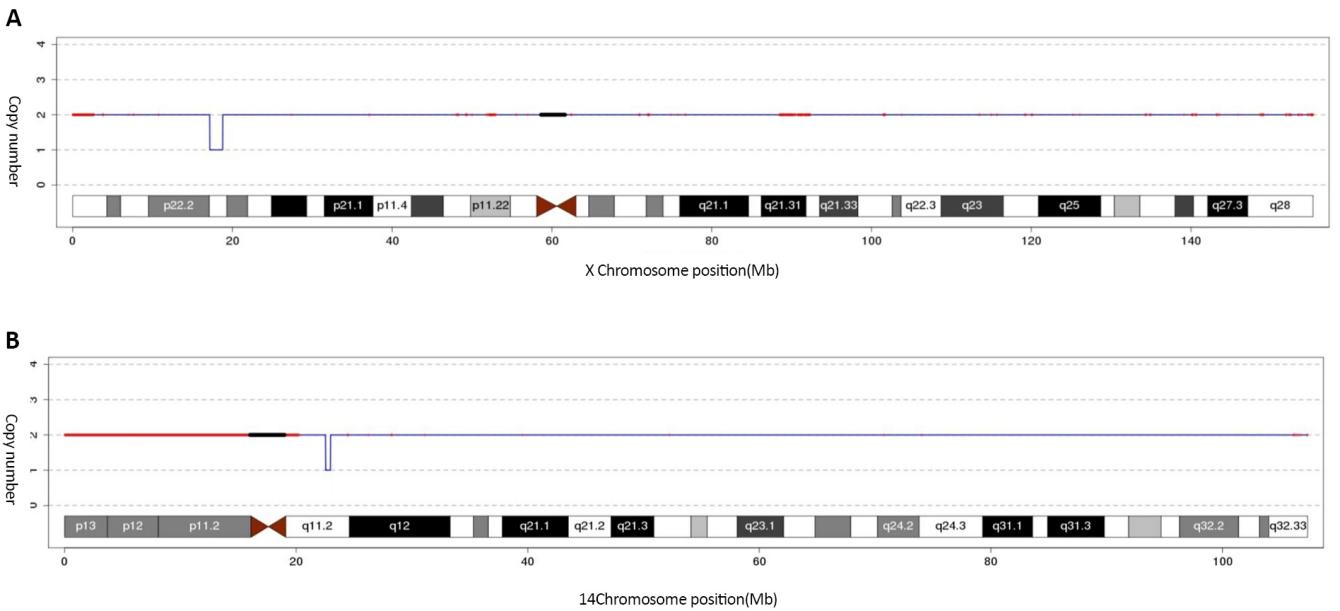
Figure 3. The identification of 1.62Mb at Xp22.13 (17160001-18780000) (GRCh37/hg19) by CNV-seq showing the microdeletion (A) and an interstitial microdeletion at 14q11.2 was identified by CNV-seq (B).
The results of fluorescence quantitative PCR were based on the ALB gene as the internal reference, and the NHS and CDKL5 genes located in the Xp22.13 region were selected for primer design. Normal control samples and child and parents samples were tested by fluorescence quantitative PCR in the same group. Consequently, the NHS and CDKL5 genes to normal control copy number ratio was about 0.5, with loss of heterozygosity. The copies of the genes in this region were normal in both parents, suggesting that the chromosomal deletion was new (Figure 4).
|
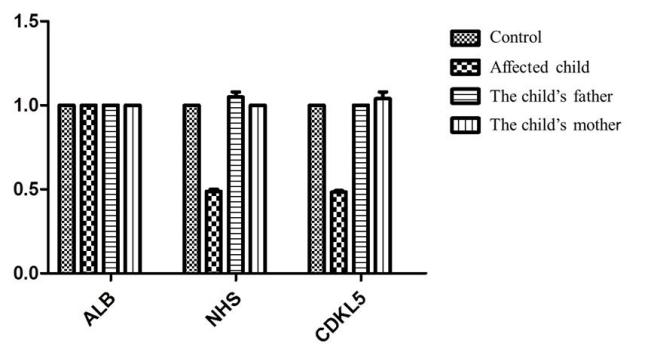
Figure 4. QF-PCR result showing heterozygous deletion of NHS and CDKL5 genes in the affected subject.
5 DISCUSSION
Genetic factors account for 40% of developmental delay/metal disability, of them, chromosomal anomalies occupy 25%[8]. G-banding karyotype analysis is a routine clinical cytogenetic diagnosis method. But <5Mb duplications and deletions cannot be detected even with high-resolution G-banding. Detection rate of conventional G-banding techniques for mental retardation, organ malformation, and growth retardation caused by a relatively small range of chromosomal imbalance aberrations is only 15% to 40%, and the missed diagnosis rate is as high as 60% to 85%[9,10]. High throughput sequencing technology can detect chromosomal aberrations and gene CNVs at the submicroscopic level with high resolution, high throughput and high accuracy, which improves the diagnostic efficiency of genetic diseases.
The balanced translocation of this case was inherited from the mother, which did not lead to the deletion and duplication of clinically significant genetic material. Therefore, balanced translocation is not the cause of a series of clinical manifestations. Molecular genetic testing found that there was a 1.62mb deletion at p22.13 on the X chromosome of the child, including NHS, CDKL5, Scm polycomb group protein like 2 (SCML2), RS1, Scm polycomb group protein like 1 (SCML1), REPS2, RAI2 genes. According to Van Esch et al.[11] and Mathys et al.[12], 2 distinct male children showing close phenotypes were identified, and they carried the 2,8-MB microdeletion on Xp22.13. The two microdeletions contain CDKL5 and NHS genes. It was worth noting that such cases exhibited innate cataract in both eyes, epileptic encephalopathy, dental abnormalities and Fallot tetralogy. Liao et al.[13] and Accogli et al.[14] described three brothers developing NHS who carried one microdeletion on Xp22.13. Apart from characteristic NHS phenotype, these patients exhibited global developmental retardation and hypotonia. The main manifestations of our patient were congenital cataract, special facial features (narrow and long face, prominent nose, etc.), abnormal teeth, intellectual disability, nystagmus, finger deformities, etc., which were consistent with NHS. At the same time, the patient also had severe early-onset intractable epilepsy, no speech function, severe psychomotor retardation, hypotonia, etc. Notably, our patient was a girl, which was rare in previous reports.
After querying ClinGen database resources, pathogenicity of insufficient haploid doses of NHS [ISCA-35637] and CDKL5 genes [ISCA-14810] was supported (haploinsufficiency score: 3). Khan et al.[6] found the new mutation on NHS (p.Lys744AsnfsX15 [c.2232delG]) from 7 cases developing infantile or innate cataract, such as long face, bulbar nose and dentition abnormalities, and the mutation could also be seen among 4 asymptomatic female children who had Y-centered lens opacities, rather than in additional 2 asymptomatic females or 3 males with clear lenses. Using whole exome sequencing, Tian et al.[15] found that NM_198270: c.1045 + 2T >A, a donor splicing site mutation in NHS gene was the pathogenic mutation for the NHS family, carrier females had no obvious abnormality in intelligence, facial features, nystagmus, microcornea, strabismus and high myopia, while cataract was not seen because of cataract surgery before this work. One female carrier, who carried c.263_266delCGTC (p.Ala89TrpfsTer106), the new NHS gene small deletion, had bilateral posterior Y-suture lens opacities and slight dental anomalies[16]. Miller et al.[17] described NHS gene disruption because of balanced translocation t(X;19)(Xp22.13;q13.1), which was the specific pathogenesis of NHS referred NHS gene’s dose effects on phenotype expression and manifestation, besides, female carriers showed congenital cataracts and Glaucoma. Maortua et al.[18], Bahi-Buisson et al.[19] and other authors[20,21], who associate CDKL5 gene mutations with various overlapping phenotypes, from autism and mental retardation to RTT with epilepsy phenotypes. Mei et al.[22] also showed that CDKL5 gene deletion/duplication may be the main cause of girls' early refractory seizures (unknown etiology).
Therefore, the clinical manifestations of the patient in this study may be related to the haploinsufficiency of NHS and CDKL5 gene, there is no additional gene, which can possibly affect the affected girl health and clinical manifestations. Retinoic acid (RA) participates in regulating early embryogenesis and cell differentiation, while RAI2 gene deletion has been elucidated that, the decreased RA signaling induces early cardiac defects in diverse animal models as well as mammalian embryos, and also lead to intellectual impairment[23]. REPS2 gene shows up-regulation within brain tissues and is associated with signaling pathway that involves small GTPases in Rho family, haploinsufficienc of REPS2 gene may also lead to mental retardation[13]. The SCML1 gene was preferentially located within testicular germ stem cells, which regulates the ubiquitination of histone H2A to establish male germline epigenome[24], so the impact on girls may be small. According to map location and homology analysis, SCML2 was the possible gene related to Xp22-linked developmental disorders, like oral-facial-digital type I syndrome[25]. According to Huopaniemi et al.[26], RS1 gene expression levels within different uterine and placental cells were analyzed, as a result, apart from retina, RS1 expression could be detected within the uterus. Consequently, RS1 protein may be related to embryonic survival and implantation.
6 CONCLUSION
Clinical manifestations caused by Xp22.13 microdeletion are complex and diverse. In the future, more investigations should be conducted for defining the Xp22.13 microdeletion phenotypic range, and the appropriate gene functions at the specific locus should be specified.
Acknowledgements
We thank the patients and their families for their participation in this study. This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.01-2019.316.
Conflicts of Interest
The authors declared that there is no conflict of interest regarding the publication of this paper.
Author Contribution
Guo Q and Sun DL designed the overall research strategy and supervised the whole process. Zhang J and Wang K recruited the case and performed the prenatal counseling. Chen WQ carried out sampling and part of the genetic experiments. Zhang J and Peng YY performed the other part of the genetic experiments, chromosome analysis and data analysis. Zang J and Sun DL together prepared this manuscript.
Abbreviation List
CDKL5, Cyclin-dependent kinase-like 5
CNV, Copy number variation
CNVseq, CNV sequencing
gNDA, Genomic DNA
NHS, Nance-Horan syndrome
PCR, Polymerase chain reaction
RAI2, Retinoic acid induced 2
REPS2, RALBP1 associated Eps domain containing 2
RS1, Retinoschisin1
SCML1, Scm polycomb group protein like 1
SCML2, Scm polycomb group protein like 2
References
[1] Nance WE, Warburg M, Bixler D et al. Congenital X-linked cataract, dental anomalies and brachymetacarpalia. Birth Defects Orig Artic Ser, 1974; 10: 285-91.
[2] Francis PJ, Berry V, Hardcastle AJ et al. A locus for isolated cataract on human Xp. J Med Genet, 2002; 39: 105-109. DOI: 10.1136/jmg.39.2.105
[3] Burdon KP, McKay JD, Sale MM et al. Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am J Hum Genet, 2003; 73: 1120-1130. DOI: 10.1086/379381
[4] Tug E, Dilek NF, Javadiyan S et al. A Turkish family with Nance-Horan Syndrome due to a novel mutation. Gene, 2013; 525: 141-145. DOI: 10.1016/j.gene.2013.03.094
[5] Migeon BR. X-linked diseases: susceptible females. Genet Med, 2020; 22: 1156-1174. DOI: 10.1038/s41436-020-0779-4
[6] Khan AO, Aldahmesh MA, Mohamed JY et al. Phenotype-genotype correlation in potential female carriers of X-linked developmental cataract (Nance-Horan syndrome). Ophthalmic Genet, 2012; 33: 89-95. DOI: 10.3109/13816810.2011.634881
[7] Craig CP, Calamaro E, Fong CT et al. Diagnosis of FOXG1 syndrome caused by recurrent balanced chromosomal rearrangements: Case study and literature review. Mol Cytogenet, 2020; 13: 40. DOI: 10.1186/s13039-020-00506-1
[8] Miclea D, Peca L, Cuzmici Z et al. Genetic testing in patients with global developmental delay / intellectual disabilities. A review. Clujul Med, 2015; 88: 288-292. DOI: 10.15386/cjmed-461
[9] Moeschler JB, Shevell M, Committee on Genetics et al. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics, 2014; 134: e903-e918. DOI: 10.1542/peds.2014-1839
[10] Hultén MA, Dhanjal S, Pertl B. Rapid and simple prenatal diagnosis of common chromosome disorders: advantages and disadvantages of the molecular methods FISH and QF-PCR. Reproduction, 2003; 126: 279-97. DOI: 10.1530/rep.0.1260279
[11] Van Esch H, Jansen A, Bauters M et al. Encephalopathy and bilateral cataract in a boy with an interstitial deletion of Xp22 comprising the CDKL5 and NHS genes. Am J Med Genet, 2007; 143: 364-369. DOI: 10.1002/ajmg.a.31572
[12] Mathys R, Deconinck H, Keymolen K et al. Severe visual impairment and retinal changes in a boy with a deletion of the gene for Nance-Horan syndrome. Bull Soc Belge Ophtalmol, 2007; 305: 49-53.
[13] Liao HM, Niu DM, Chen YJ et al. Identification of a microdeletion at Xp22.13 in a Taiwanese family presenting with Nance-Horan syndrome. J Hum Genet, 2011; 56: 8-11. DOI: 10.1038/jhg.2010.121
[14] Accogli A, Traverso M, Madia F et al. A novel Xp22.13 microdeletion in Nance-Horan syndrome. Birth Defects Res, 2017; 109: 866-68. DOI: 10.1002/bdr2.1032
[15] Tian Q, Li Y, Kousar R et al. A novel NHS mutation causes Nance-Horan Syndrome in a Chinese family. BMC Med Genet, 2017; 18: 2. DOI: 10.1186/s12881-016-0360-9
[16] Li H, Yang L, Sun Z et al. A novel small deletion in the NHS gene associated with Nance-Horan syndrome. Sci Rep, 2018; 8: 2398. DOI: 10.1038/s41598-018-20787-2
[17] Miller C, Gertsen BG, Schroeder AL et al. Allelic and dosage effects of NHS in X-linked cataract and Nance-Horan syndrome: A family study and literature review. Mol Cytogenet, 2021; 14: 48. DOI: 10.1186/s13039-021-00566-x
[18] Maortua H, Martínez-Bouzas C, Calvo MT et al. CDKL5 gene status in female patients with epilepsy and Rett-like features: Two new mutations in the catalytic domain. BMC Med Genet, 2012; 13: 68. DOI: 10.1186/1471-2350-13-68
[19] Bahi-Buisson N, Nectoux J, Rosas-Vargas H et al. Key clinical features to identify girls with CDKL5 mutations. Brain, 2008; 131: 2647-2661. DOI: 10.1093/brain/awn197
[20] Mari F, Azimonti S, Bertani I et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet, 2005; 14: 1935-1946. DOI: 10.1093/hmg/ddi198
[21] Psoni S, Willems PJ, Kanavakis E et al. A novel p.Arg970X mutation in the last exon of the CDKL5 gene resulting in late-onset seizure disorder. Eur J Paediatr Neurol, 2010; 14: 188-191. DOI: 10.1016/j.ejpn.2009.03.006
[22] Mei D, Marini C, Novara F et al. Xp22.3 genomic deletions involving the CDKL5 gene in girls with early onset epileptic encephalopathy. Epilepsia, 2010; 51: 647-654. DOI: 10.1111/j.1528-1167.2009.02308.x
[23] Stefanovic S, Zaffran S. Mechanisms of retinoic acid signaling during cardiogenesis. Mech Dev, 2017; 143: 9-19. DOI: 10.1016/j.mod.2016.12.002
[24] Hasegawa K, Sin HS, Maezawa S et al. SCML2 establishes the male germline epigenome through regulation of histone H2A ubiquitination. Dev Cell, 2015; 32: 574-588. DOI: 10.1016/j.devcel.2015.01.014
[25] Montini E, Buchner G, Spalluto C et al. Identification of SCML2, a second human gene homologous to the Drosophila sex comb on midleg (Scm): A new gene cluster on Xp22. Genomics, 1999; 58: 65-72. DOI: 10.1006/geno.1999.5755
[26] Huopaniemi L, Tyynismaa H, Rantala A et al. Characterization of two unusual RS1 gene deletions segregating in Danish retinoschisis families. Hum Mutat, 2000; 16: 307-314. DOI: 10.1002/1098-1004(200010)16:4<307::AIDHUMU3>3.0.CO;2-L
Copyright © 2023 The Author(s). This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.


