Anti-angiogenic Therapy for Tumor: Tumor Angiogenesis, Vasculogenic Mimicry and Vascular Normalization
Dandan Du1, Fulian Gong1,2, Wanying Zhang1, Bingfang Yu1, Xiuli Guo1*
1Department of Pharmacology, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, Jinan, Shandong Province, China
2Department of Pharmacy, Tianjin Children’s Hospital, Tianjin, China
*Correspondence to: Xiuli Guo, PhD, Professor, Department of Pharmacology, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, No. 44 Wenhua West Road, Jinan 250012, Shandong Province, China; Email: guoxl@sdu.edu.cn
Abstract
The growth and metastasis of solid tumors require a sufficient blood supply that provides oxygen and nutrients. In recent years, targeting tumor vasculature has gained rapid development, owing to the abnormalities of tumor blood vessels compared with normal blood vessels. There are three aspects for targeting blood vessels in tumor treatment, including angiogenesis, vasculogenic mimicry (VM) and vascular normalization. Anti-angiogenic therapy is an anti-tumor strategy targeting to the new blood vessels. VM is an alternative form of blood supply not depending on endothelial vessels, but insteading to form a tubular structure similar to blood vessels by the tumor cells themselves. However, anti-angiogenic therapy may have some limits such as tumor hypoxia, reduction of chemotherapy drugs approaching to tumors, etc. As a different approach to anti-tumor therapy, vascular normalization provides a new idea for the cancer treatment. In this review, we summarized the advanced researches on therapies of anti-angiogenic, anti-VM and vascular normalization, including their molecular mechanisms and clinical significances.
Keywords: tumor angiogenesis, vasculogenic mimicry, vascular normalization, anti-angiogenic therapy
1 INTRODUCTION
Targeting tumor vasculature as a means to inhibit tumor growth and metastasis and its beneficial effects are widely acknowledged recently[1]. Tumor cells need to develop new blood vessel networks to maintain their rapid proliferation. It is widely accepted that a solid tumor cannot grow further than approximately 2mm3 without blood vessels that provide oxygen and nutrients[2]. However, compared with normal blood vessels, the morphology and function of tumor blood vessels are different and abnormal[3]. In tumors, tumor cells, stromal cells and other cells in the microenvironment can secret high levels of growth factors causing tumor vascular development to be abnormal of which vascular endothelial growth factor (VEGF) is a critical one[4]. The growth factors could promote the formation of abnormal blood vessels, and these poorly functioning vessels can also have an effect on the tumor microenvironment, such as local hypoxia, decreased immune cell function and tumor metastasis[5,6]. At present, there are two main anti-tumor strategies for tumor blood vessels, one is to inhibit angiogenesis, and the other is to directly destroy the existing tumor vasculature[4].
Unlike the classic mechanisms of tumor angiogenesis, VM is an alternative form of blood supply not depending on endothelial vessels. In 1999, VM was introduced firstly in highly aggressive melanoma cells that obtained endothelial-like characteristics[7]. VM, as a tumor-specific angiogenesis, has been observed in a variety of highly invasive tumors and is considered as a novel model for neoangiogenesis in invasive tumors. The blood vessels formed in VM consist of an array of endothelial tumor cells supported by periodic acid-Schiff (PAS) positive cells and abundant external stromal components. The wall of the VM channel was positive for PAS staining, while the outside lumen rich in tumor cells was negative for CD31 or CD34 staining[8]. These tubes transport nutrients and red blood cells carrying oxygen to the tumor. VM, as a new target for tumor vasculature, was proposed as a new model for tumor perfusion, including sarcomas, carcinomas, glioblastomas, astrocytomas, melanomas, etc. VM can also form a connection with blood vessels, further exacerbating the tumor’s metastasis via blood circulation[9]. There are multiple potential mechanisms for VM formation, such as EMT and cancer stem cells (CSCs)[10], and various signaling pathways that contribute to VM formation, including VEGF, phosphatidylinositol 3-kinase (PI3K), vascular endothelial cadherin (VE-cadherin), erythropoietin induced hepatocyte receptor A (EphA2), hypoxia inducible factor (HIF-1α), etc[11,12]. As a tumor specific angiogenesis, VM has been shown to enhance metastasis after anti-angiogenesis therapy[13]. Therefore, studies in the VM field will contribute to improving cancer treatment methods.
Vascular normalization is different from the traditional anti-vascular therapies[14]. This strategy aims to reduce the number of abnormal blood vessels while allowing mature ones with pericyte-covered and intact basement membranes, resulting in restoring proper tumor perfusion and oxygenation. Normalization of the tumor vascular system restores normal vascular function and may help in preventing cancer cells from acquiring the aggressive phenotypes associated with hypoxic microenvironment. In addition, increasing tumor perfusion can improve the efficacy of chemotherapy, radiotherapy and immunotherapy[15]. Therefore, vascular normalization might limit tumor cell invasiveness and enhance the effectiveness of anticancer treatment, which has appeared as a new therapeutic strategy[16].
Either tumor angiogenesis that is dependent on endothelial cells or VM that is dependent on tumor cells is closely related to the development of cancer. The effect of anti-vascular therapies and vascular normalization is regulated by many factors, including the hypoxic microenvironment, the interaction of tumor cells with the surrounding environment, and the remodeling of extracellular matrix[3,17]. Therefore, understanding the biological characteristics and formation mechanisms of tumor vasculature and signaling pathways and normalization of tumor blood angiogenesis may offer a theoretical basis for cancer treatment. In this review, we will introduce molecular mechanisms that influence tumor angiogenesis, VM and vascular normalization that might provide a possible new targeting strategy for cancer therapy.
2 THE ABNORMALITY OF TUMOR BLOOD VESSELS
In comparison with normal blood vessels in human, tumor blood vessels exhibit abnormal morphological and biological characteristics. The normal vascular network has a uniform distribution of mature blood vessels to allow oxygen and nutrients to perfuse all cells. In tumors, immature blood vessels form a chaotic labyrinth in which small arteries, capillaries, and venules are difficult to distinguish. The distinction between solid tumor vasculature and normal vasculature also provides a unique target for anticancer therapy.
2.1 Abnormal Morphological Manifestations of Tumor Vessels
The most obvious feature of tumor blood vessels is their immatureness. It is characterized by vascular wall dysplasia, lack of coverage of pericytes, usually with discontinuous endothelial cell lining, poor correlation with vascular smooth muscle cells, and abnormal basement membrane[18]. Endothelial cells in tumor tissues also exhibit irregularities, and interconnected cells are loosely interconnected which could increase vascular permeability and increase interstitial pressure. In addition, the ability of tumor blood vessels to transport nutrients and eliminate metabolic waste is greatly reduced[4]. This abnormal morphological manifestation of tumor blood vessels also adversely affects the entry of anti-tumor drugs into cancer cells for cancer treatment.
2.2 Abnormal Phenotype of Tumor Blood Vessels
For many years, human umbilical vein endothelial cells have been used as study objects in the researches of tumor blood vessels, while few studies have focused on endothelial cells in the tumor microenvironment. Recent reports have shown molecular differences between tumor endothelial cells (TEC) and normal endothelial cells[19]. The cytogenetic abnormalities of TEC may be contributed to the resistance of tumors to chemotherapeutic drugs[20]. TEC is more responsive to some growth factors such as endothelial growth factor and VEGF. Tumor cells, tumor stromal cells, and TEC could secrete large amounts of VEGF, which act on the corresponding receptor on the TEC in an autocrine or paracrine manner to help the TEC form a pro-angiogenic phenotype and an anti-apoptotic phenotype[21].
2.3 Heterogeneity of TEC
TEC have been found to be highly heterogeneous[22,23]. In tumors, the morphology of blood vessels and the coverage of pericytes vary with the degree of tumor malignancy. With the increasing of malignancy, the blood vessels become more immature and the coverage of pericytes becomes lower[24]. Some genes involved in angiogenesis are highly expressed in TEC, such as genes encoding VEGF/VEGFR2 and PI3K/AKT signaling pathways. TEC also express matrix metalloproteinases (MMPs) to degrade the basement membrane[25,26]. Under hypoxic conditions, tumor cells can promote the expression of certain growth factors through genetic alterations to further induce metabolic adaptation and promote angiogenesis[27]. All of the above genetic changes of TEC contribute to tumor progression and metastasis.
2.4 Abnormal Biological Characteristics of Tumor Vessels
Irregularities of vascular networks increase the geometric resistance of the blood flow, so a small decrease in perfusion pressure that affects normal tissue may have a large effect on tumor tissue[28]. Therefore, tumor blood vessels have abnormal rheology. The high leakage of tumor blood vessels also makes it difficult for chemotherapeutic drugs to enter cells. In tumor tissue, capillaries contain high levels of deoxygenated blood, causing local hypoxia in solid tumors. However, the tumor itself can adapt to this unfavorable growth environment through a series of genetic changes, such as metabolic adaptation and promotion of angiogenesis. The hypoxic state of tumors can also lead to resistance to anti-tumor treatment, of which the resistance to immunotherapy is important[29,30].
3 ALTERNATIVE NEOVASCULARIZATION PROCESS IN TUMORS
In addition to sprouting of angiogenesis and angiogenesis involved in endothelial progenitor cells, there are some alternative methods that allow solid tumors to evade VEGF-targeted therapy[31,32]. Here we introduce several important alternative angiogenesis methods including the co-option of blood vessels, vascular remodeling, and VM that is dependent on tumor cells[33]. Exploring these alternative angiogenic mechanisms not only complements our understanding of tumor angiogenesis, but also helps overcome the resistance of vascular therapies.
3.1 Vessel Co-option
In the tumor tissues rich in vascular networks, new blood vessels can be formed by vessel co-option[34]. The occurrence of blood vessel co-option is closely related to the site of its occurrence. In vascular-rich tissues, cancer cells recruit already existing blood vessels in the surrounding normal tissues, and this process does not involve the generation of new blood vessels. Then cancer cells grow along existing blood vessels[35]. This process is more common in highly aggressive tumors.
The co-option blood vessels are usually supported by pericytes from the periphery[36]. In the models of human ovarian cancer and esophageal cancer treated with bevacizumab, the density of pericytes surrounding vessels increases[37]. In a murine brain melanoma model for analyzing the interactions of human melanoma cells with microvascular channels, a striking spread of melanoma cells along preexisting microvascular channels was emerged with features of both vascular co-option and angiotropism/pericytic mimicry[38]. The above results suggest that continuous vascular co-option is one of the main mechanisms by which tumors escape anti-angiogenic therapies.
3.2 Vessel Remodeling
The co-option of blood vessels is accompanied by the occurrence of vessel remodeling. Tumor-induced vascular remodeling provides better support for the growth of primary tumors. During vessel remodeling, some changes occur, including expanded diameter of the extravascular lumen before the co-option, and the layer of pericytes become less until it disappears, which promotes the exchange of oxygen and nutrients between the tumor tissue and blood circulation[39]. Studies have reported that vascular remodeling occured in metastatic lymph nodes in high endothelial venules[40]. Moreover, in metastatic cancer cells, the remodeling blood vessels could be further incorporated into the tumor vasculature. The occurrence of vascular remodeling exacerbates the therapeutic resistance caused by vessel co-option.
3.3 Vasculogenic Mimicry (VM)
The VM, an endothelial cell-independent angiogenesis, has been found in some highly invasive tumor cells, such as human melanoma, liver cancer, double differentiated tumors, etc[9]. These tumor cells have the ability of endothelial cells to form a bureaucratic cavity and are interconnected to form a simulated blood vessel. VM can also be combined with channels formed by vascular endothelial (VE) cells to provide blood to tumor cells. In fact, during the formation of VM, tumor cells express vascular markers represented by VE-cadherin that plays an important role in VM. Moreover, the formation of VM also helps cancer cells to enter the bloodstream and metastasize, exacerbating cancer recurrence and poor prognosis[41].
4 SIGNALING MOLECULES IN TUMOR ANGIOGENESIS AND VM
During the complex process of tumor vasculature formation, both the angiogenesis that depends on endothelial cells and VM depending on tumors could be regulated by various factors in the tumor microenvironment. However, the mechanisms of tumor angiogenesis and VM are both similar and different. The following is a description of the signal transduction pathways involved in the formation of these two type blood vessels.
4.1 VEGF Signaling in Tumor Angiogenesis
VEGF has been identified as having endothelial cell-specific mitogens that induce physiological and pathological angiogenesis. Members of this family include VEGFA, VEGFB, VEGFC, VEGFD, and placenta derived growth factor[25]. Due to alternative cleavage, functionally, VEGF has several distinct variants, namely VEGF121, VEGF145, VEGF148, VEGF165, VEGF183 and VEGF189[42]. The role of VEGF in angiogenesis and lymphangiogenesis has been studied extensively and provides the basis for anti-angiogenic therapies that target VEGF and its receptors.
VEGF interact with corresponding receptors on VE cells by autocrine or paracrine. The classical VEGF receptors are the tyrosine kinase receptors (RTKs) which consist of VEGFR1, VEGFR2 and VEGFR3[43]. As the main functional receptor of VEGF, VEGFR2 can autophosphorylate upon binding to VEGF, which in turn activates downstream signaling to form a cascade that affects proliferation, survival, migration and permeability of endothelial cell. VEGF transduction pathways in VE cells are shown in Figure 1. The specific mechanisms of related proteins included PLC-ERK1/2, SRC-FAK, PI3K-AKT, etc.
|
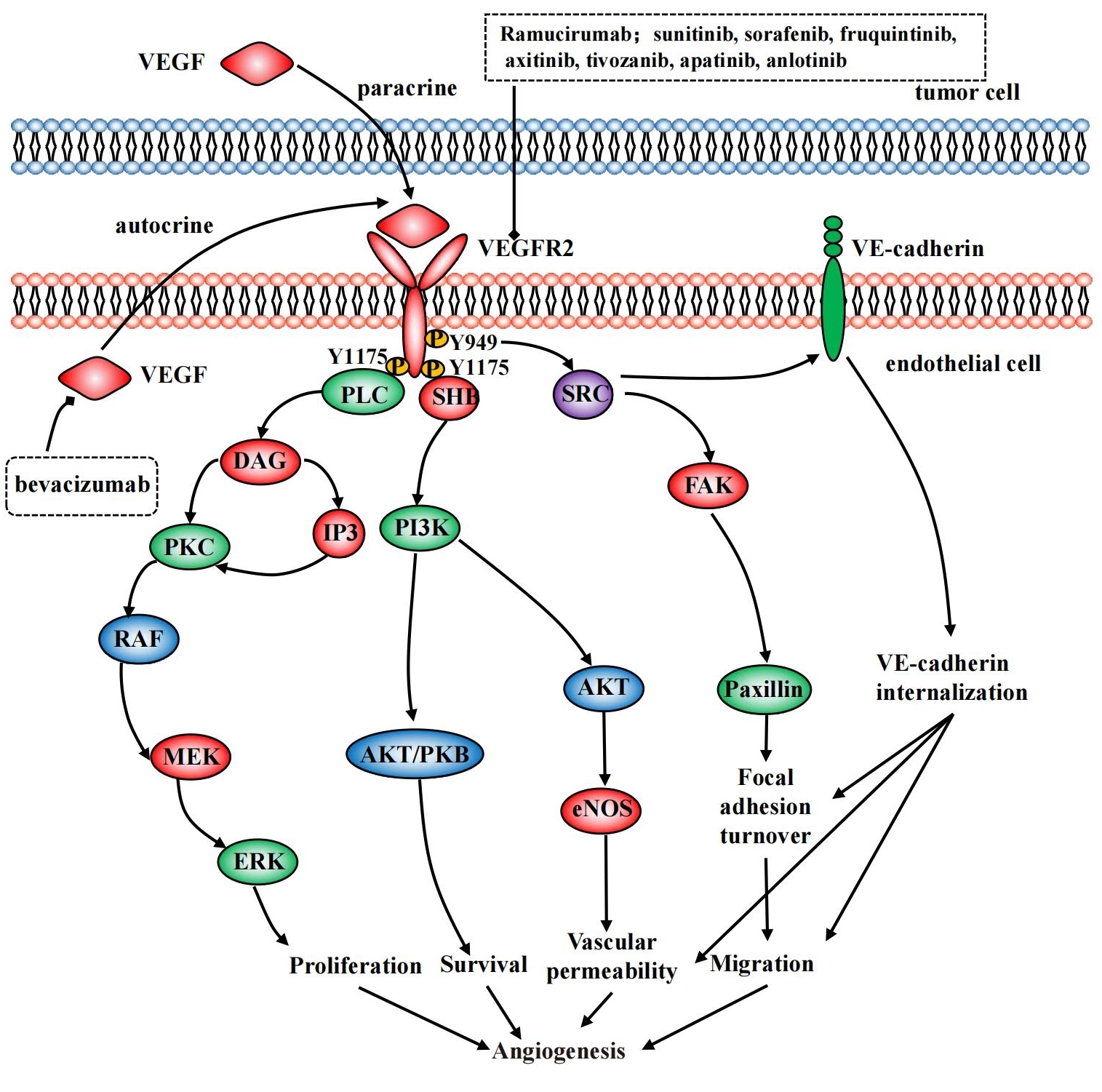
Figure 1. VEGF transduction pathways and VEGFR inhibitors in VE cells.
4.1.1 PLC-ERK1/2
Phosphorylation of VEGFR2 Y1175 in human proteins activates phospholipase Cγ and then both inositol 1,4,5-triphosphate and diacylglycerol (DAG) can be produced. Afterwards protein kinase C could be activated by DAG and this action can promote endothelial cell proliferation through the RAF1-MEK-ERK1/2 signaling cascade. This process plays an important role in human vascular development and adult arterial development[44].
4.1.2 SRC-FAK
Activation of SRC by VEGF-A/VEGFR2 is dependent on Y949 in the receptor kinase insert[45,46]. After the activation of SRC family kinase, VE-cadherin phosphorylates which resulting in increased vascular permeability. In addition, the downstream focal adhesion kinase (FAK) and paxillin can also be activated further affecting permeability and adhesion of endothelial cells.
4.1.3 PI3K-AKT
PI3K is an important regulator of angiogenesis. Under normal conditions, activated PI3K can produce lipid products as second messengers that activate target proteins within the cell, forming signaling cascades ultimately regulating cell proliferation, differentiation, survival and migration. PI3K can be directly coupled to the pY1175 site of VEGFR2, indirectly via FAK, SRC, etc. The AKT serine/threonine kinase AKT1-3 is a signaling protein downstream of PI3K with a broad substrate and affects many biological processes including cell survival, proliferation and apoptosis[47,48].
Interestingly, some tumors appear to respond to VEGF independently of VEGFR2, suggesting that other receptors might also mediate VEGF signaling[49]. Recent studies have found that neuropilin (NRP) might be a receptor for the action of VEGF. NRP functions primarily as co-receptors because they lack of the inherent signaling capabilities. For example, NRP forms complexes with specific neurites of neurons and other cell types to act as a functional semaphore receptor[50,51].
In addition to being an important player in inducing angiogenesis, VEGF has other functions in the tumor microenvironment. It is worth noting that the function of immune cells is regulated by VEGF[25]. Studies found that VEGF could promote the chemotaxis of various immune cells to tumor tissues through autocrine, including myeloid-derived suppressor cells, regulatory T cells, dendritic cells and macrophages, and subsequently reduce the body’s immune response. At the same time, the expression of VEGFR2 in immune cells is also a focus of future research, but its specific molecular mechanism needs further study[52].
4.2 HIF-1α, c-Jun and Angiogenesis
The limited blood supply within the tumor tissue can lead to hypoxia in rapidly proliferating tumor cells, which may further worsen the degree of hypoxia in solid tumors. Hypoxia in the tumor leads to stabilization of HIF family. HIF is a heterodimeric transcription factor composed of HIF-α (HIF-1α, HIF-2α, and HIF-3α) and HIF-β (HIF-1β, HIF-2β and HIF-3β). HIF-α is a functional subunit that determines the activity of HIF. HIF-β is a structural subunit that can be stably expressed in cells. Under normoxic conditions, HIF-α binds to the Von Hippel-Lindau (VHL) gene product pVHL, and is then recognized by the E3 ubiquitin-binding enzyme receptor to form a complex, which is further degraded by the proteasome. This process also requires the involvement of oxygen-dependent prolyl hydroxylase. However, under hypoxic conditions, pVHL loses its activity and HIF-α can be stably expressed in cells[27,53]. Tumor cells can undergo a series of genetic changes to adapt to the hypoxic microenvironment. One of the key downstream targets of the HIF pathway is VEGF[54]. Zhao et al.[55] found that inhibition of HIF-1a prevented endothelial cell angiogenesis. In addition, HIF-2α has also been shown to be involved in tumor angiogenesis. Skuli et al.[56] found that HIF-2α deficiency in mouse EC reduced vascular function and tumor angiogenesis.
In addition, HIF-1α also protects itself from degradation by c-Jun, which binds to the HIF-1α oxygen-dependent degradation domain in a non-transcribed manner[57]. c-Jun is a common transcription factor relative with cell proliferation, differentiation, apoptosis, oxidative stress and the like. Studies have shown that under hypoxic conditions, c-Jun forms a complex with HIF-1α to regulate VEGF expression and transcription of multidrug resistance MDR1 gene through JNK phosphorylation. Moreover, c-Jun can also be activated by HIF-1α[58]. Up-regulation of VEGF expression induced by HIF-1α can further activate the VEGF/VEGFR2 signal transduction pathway and initiate angiogenesis in tumor tissues. Although there have been some reports on the mutual regulation of HIF-1α and c-Jun, the specific regulation mechanism and its influence on the development of tumor angiogenesis needs further clarification.
4.3 VE-cadherin, EphA2, FAK and VM
VE-cadherin, as one member in cadherin family, is specifically expressed in endothelial cells[59]. Studies have shown that VE-cadherin was an important participant in the formation of VM in melanoma. The deletion of VE-cadherin in melanoma does not form a complete VM[60]. EphA2, a tyrosine kinase receptor that is overexpressed in highly aggressive melanoma, can induce the re-localization of VE-cadherin in the cell membrane. This co-localization of two proteins further promotes the phosphorylation of EphA2, which subsequently leads to phosphorylation of FAK. In addition, EphA2 also promotes the expression of MMP2, which is likely to be dependent on phosphorylation of FAK. Reports have shown that the formation of VM is interrupted when FAK is inhibited. FAK is a tyrosine kinase involved in various biological processes of cells, such as cell invasion, migration, adhesion and so on[9]. After the above-mentioned VM molecular model was identified, Hendrix et al.[16] proposed a signal transduction pathway for designing VM of tumor cells. Activated FAK can further trigger the activation of PI3K, which in turn regulates the formation of VM. Moreover, the co-localization of VE-cadherin and EphA2 can directly activate FAK that is independent on PI3K[59]. Similar to the angiogenesis of endothelial cells, the forehead formation of VM is also regulated by HIF. In the hypoxic environment, the VM formation increases, however, the initial initiator of VM in this pathway remains unknown.
4.4 Nodal, Notch and VM
CSC refers to cancer cells with the characteristics of stem cells, that is, self-replication and multi-cell differentiation. The stemness of tumor cells is closely related to their plasticity. Two signaling pathways, Nodal and Notch, are the major signaling pathways associated with cell stemness[61]. Generally speaking, the highly invasive tumors cells have increased stemness and enhanced plasticity.
Nodal is usually not present in normal human tissues, but can be over-activated in tumor tissues. Recent studies have found that Nodal was highly expressed in a variety of cancers, including breast cancer, colon cancer, testicular cancer, leukemia, prostate cancer, colon cancer, glioma and neuroblastoma. Nodal has been shown to promote the formation of VM structures. Inhibition of Nodal with inhibitors or neutralizing antibodies significantly reduced the ability of tumor cells to form VM[62,63].
In addition to Nodal, the highly conserved Notch signaling pathway, which is usually activated by a ligand on adjacent cells (DLL1/2/4 or JAG1/2), plays an important role in cell differentiation and cell-renewal. Thereby, targeting Notch/Nodal signaling pathway is an effective strategy for inhibiting the formation of VM in anti-tumor therapies[61,64]. The signaling molecules implicated in tumor cell VM were overviewed in Figure 2.
|
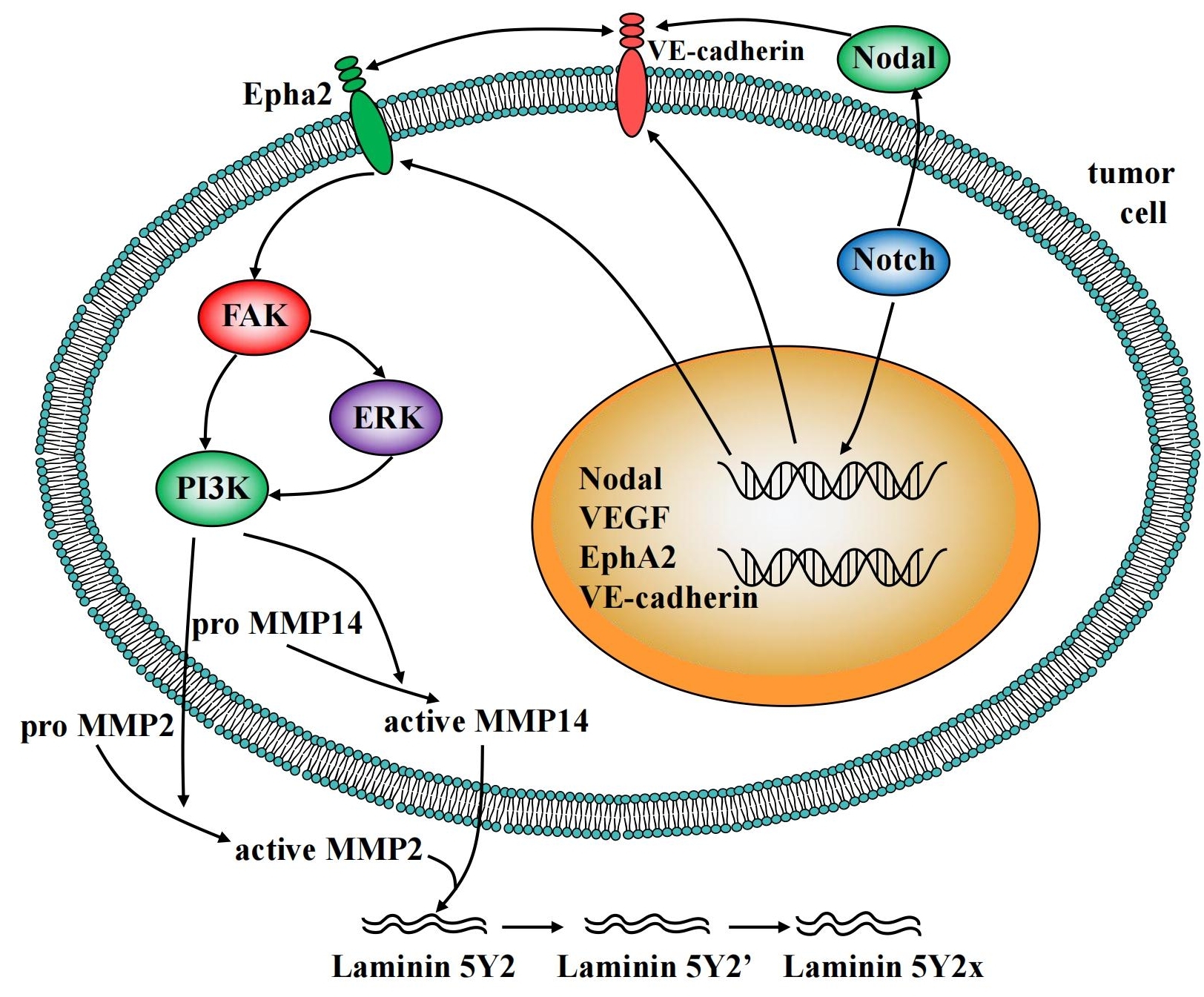
Figure 2. Overview of the signaling molecules implicated in tumor cell vascular mimicry.
5 THERAPIES TARGETING TUMOR VASCULATURE
Since the Food and Drug Administration (FDA) approved bevacizumab (the first angiogenesis inhibitor) for the treatment of metastatic colorectal cancer, angiogenesis has become an attractive target for cancer treatment. A large number of angiogenesis inhibitors have been developed, including monoclonal antibodies, endogenous peptides and small molecule targeted drugs. Moreover, in recent years, great progress has been made in the normalization of blood vessels.
5.1 Protein/Peptide Angiogenesis Inhibitors
5.1.1 Proteins
In VEGF targeting therapies, monoclonal antibodies have been used as important agents for tumor treatment, of which bevacizumab possesses the most focus for many tumor types preventing VEGF from binding to receptors and activating the signaling cascade that finally leads to angiogenesis[65]. Moreover, bevacizumab can also be used in combination with conventional chemotherapy to improve the therapeutic effect of tumors without increasing toxicity except metastatic colorectal cancer. In non-small cell lung cancer, bevacizumab can improve the efficacy of paclitaxel and carboplatin[66].
In addition to bevacizumab, other antibodies targeting VEGF signaling are also being tested. Ramucirumab, a monoclonal antibody targeting VEGFR2[67], which has strong inhibitory effects on gastric cancer, non-small cell lung cancer and metastatic colorectal cancer, and exhibits similar pharmacokinetics, were approved by FDA in 2014. Furthermore, studies of new neutralizing antibodies against different epitopes of human VEGF have shown that epitope disturbing selection with a triple mutant selector antigen was very useful to block anti-VEGF antibody fragment against neutralizing epitope on human VEGF[68]. VA12 as a nanobody targeting VEGFA has been developed, which has higher tissue penetration[69].
5.1.2 Peptides
The study of bioactive endogenous amino acid fragments inhibiting microvascular formation provides the basis for peptides in anti-tumor angiogenesis. Anti-angiogenic peptides are designed to bind to specific proteins in cancers, such as VEGF, VEGFR2, chemokines, etc[70]. For example, the folded peptide DI-P2 based on the Annexin A2 which has anti-angiogenesis effect could inhibit angiogenesis by interfering with VEGF. The advantages of peptides include high activity and specificity, and low immune response in comparison to proteins or domains. However, shortcomings such as low bioavailability, low stability and immunogenic effects hinder their development[71]. At present, only a modified recombinant endostatin called endostar, has been approved for clinical trials in China, and its combination with chemotherapy has become the first-line treatment for advanced non-small cell lung cancer.
5.2 Small Molecule Inhibitors
Although monoclonal antibodies that inhibit tumor blood vessels have achieved great clinical success, the search for small molecule inhibitors is becoming common in cancer therapy, such as tyrosine kinase inhibitors, microtubule-targeted drugs, etc.
5.2.1 Receptor Tyrosine Kinase Inhibitors (RTKIs)
RTKIs targeting VEGF signaling pathway are important components of small molecule inhibitors. VEGFR2 inhibitors were reported firstly as small molecules inhibiting angiogenesis. These RTKIs often inhibit the activation of VEGFR2, and then block the proliferation and vessel networks formation. In addition, these inhibitors can also inhibit other RTKIs, such as PDGF receptors, EGFR, fibroblast growth factor receptors, etc. To date, inhibitors targeting VEGF and other proteins have been developing in the different stages. RTKIs, commonly used in clinical practice, include sunitinib, sorafenib, fruquintinib, axitinib, tivozanib, apatinib, anlotinib, etc[72,73]. For example, apatinib is effective for treatment of advanced gastric adenocarcinoma and advanced hepatocellular carcinoma by inhibiting VEGFR-2[74]. Anlotinib inhibits angiogenesis via suppressing VEGFR2, PDGFRβ and FGFR1 for the treatment of non-small cell lung cancer, small lung cancer and soft tissue sarcoma[75]. In addition, in the treatment of metastatic renal cell carcinoma, axitinib was superior to sorafenib, probably because it was more specific to VEGFRs[76].
In recent years, many novel inhibitors have been extracted or synthesized, such as benzoxazole substituted triazolotriazines targeting VEGFR-2, has potent anticancer effects[77]. These small molecule inhibitors have been shown to have the potential to treat solid tumors as well as anti-angiogenic properties. For example, marine derivative apratoxin S4 was active against lung, head and neck, bladder cancer by targeting RTKs with antiangiogenic activity[78].
5.2.2 Microtubule Targeting Inhibitors
Traditional microtubule-targeted drugs have been reported to possess potential anti-angiogenic effects, including paclitaxel, colchicine, vincristine, etc. Among such drugs, microtubule inhibitors acting on the colchicine binding site are most prominent in inhibiting tumor angiogenesis and anti-drug resistance[79,80]. These drugs, by combining with specific action sites, can cause microtubule depolymerization, disrupting changes in the cytoskeleton and cell morphology. Importantly, they selectively target rapidly proliferating TEC, causing regression of tumor blood vessels[81]. In addition, low doses of these drugs can also inhibit angiogenesis that is independent of its cytotoxic effects. Combretastatin phosphate is currently in clinical research and its different derivatives are also in scientific research. Other small molecule drugs targeting microtubules, such as MT189 and DHPAC, have showed significant anti-angiogenic effects[82,83].
5.3 Vascular Normalization Theory
In cancer treatment, as an alternative to anti-angiogenic therapy, a new approach to improving tumor vascular function has been proposed, namely vascular normalization. This method generally works by reducing the number of irregular, immature blood vessels in tumor tissue, while at the same time improving the coverage of the pericytes in the blood vessels and the function of the basement membrane[84]. Studies showed that the anti-angiogenic drug bevacizumab promoted tumor vascular normalization at lower concentrations and might enhance the efficacy of chemotherapy drugs at combined administration. In addition, in the treatment of pancreatic cancer and lung cancer, cilengitide at low-dose (50mg/kg/d) combined with verapamil showed the increase in blood vessel density that could lead to more effective delivery of chemotherapy drugs to the target sites[85]. Anti-angiogenic drugs such as sorafenib also have the potential to promote tumor vascular normalization. These results showed that anti-tumor angiogenesis or promotion of tumor vascular normalization might depend on the dose of used drug.
In recent studies, the combination of tumor vascular normalization and immunotherapy is also a promising anti-tumor strategy. In the tumor hypoxic microenvironment, the function of immune cells was inhibited. Therefore, improving the tumor hypoxia microenvironment through vascular normalization can effectively improve the efficacy of immunotherapy and the characteristics of hypoxia and high permeability in tumors, which is of great significance for the treatment of tumors[86,87]. However, the underlying mechanism of drugs to promote the vascular normalization is still unclear, the normalization of blood vessels is difficult to apply to clinical practice. It is important to explore new ways to regulate the tumor vasculature.
6 PERSPECTIVES
Different tumor subtypes have different responses to different drug treatments. If significant biomarkers can be screened out, personalized treatment for different tumor subtypes will help to improve the efficacy of anti-angiogenic drugs and reduce the toxicity of drugs. In addition, there are still multiple mechanisms to escape anti-angiogenic therapy, and anti-angiogenic drugs may develop resistance. Therefore, the future challenges are the prediction of efficacy biomarkers and the search for small molecules that target tumor vessels with low toxicity and high selectivity.
7 CONCLUSION
Differences between the tumor vasculature and the normal vascular system provide an opportunity to targeting tumor vascular treatment which has developed rapidly in recent years. Many multiple signaling molecules such as VEGF and HIF involved in tumor angiogenesis. Also, VE-cadherin, EphA2, FAK and other factors involved in VM. These signaling pathways provide the beneficial basis for the drugs targeting tumor angiogenesis and the development of VM. Normalization of blood vessels opens up new perspectives for tumor-targeted vascular treatment in an opposite direction. However, these therapies might have some side effects owing to the complexity of tumor angiogenesis. It is particularly important to determine the relevant mechanisms of vascular normalization and the optimal time of drug action, and how to effectively combine different anti-tumor drugs without significant toxicity.
Acknowledgements
This work was supported by Special Funds for Local Scientific and Technological Development of Shandong Province guided by the Central Government [YDZX2021023] and National Science Foundation of China Grants [82173843].
Conflicts of Interest
The authors declared no conflict of interest.
Author Contribution
Du D, Gong F were responsible for conceptualizing and writing the original manuscript. Zhang W was responsible for writing and editing. Yu B was responsible for resources. Guo X was responsible for writing, review and editing. All authors contributed to the manuscript and approved the final version.
Abbreviation List
CSC, Cancer stem cell
DAG, Diacylglycerol
EphA2, Erythropoietin induced hepatocyte receptor A
FAK, Focal adhesion kinase
FDA, Food and Drug Administration
HIF, Hypoxia inducible factor
MMPs, Matrix metalloproteinases
NRP, Neuropilin
PAS, Periodic acid-Schiff
PI3K, Phosphatidylinositol 3-kinase
RTKIs, Receptor Tyrosine Kinase Inhibitors
RTKs, Tyrosine kinase receptors
TEC, Tumor endothelial cells
VE, Vascular endothelial
VE-cadherin, Vascular endothelial cadherin
VEGF, Vascular endothelial growth factor
VHL, Von Hippel-Lindau
VM, Vasculogenic mimicry
References
[1] Marmé D. Tumor Angiogenesis: A key target for cancer therapy. Oncol Res Treat, 2018; 41: 164.[DOI]
[2] Li S, Xu H, Wu C et al. Angiogenesis in pancreatic cancer: Current research status and clinical implications. Angiogenesis, 2019; 22: 15-36.[DOI]
[3] Hida K, Maishi N, Annan DA et al. Contribution of tumor endothelial cells in cancer progression. Int J Mol Sci, 2018; 19: 1272.[DOI]
[4] Ciesielski O, Biesiekierska M, Panthu B et al. The epigenetic profile of tumor endothelial cells. Effects of Combined Therapy with Antiangiogenic and Epigenetic Drugs on Cancer Progression. Int J Mol Sci, 2020; 21: 2606.[DOI]
[5] Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science, 2016; 352: 175-180.[DOI]
[6] Damgaci S, Ibrahimhashim A, Enriqueznavas PM et al. Hypoxia and acidosis: immune suppressors and therapeutic targets. Immunology, 2018; 154: 354-362.[DOI]
[7] Maniotis AJ, Folberg R, Hess A et al. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am J Pathol, 1999; 155: 739-752.[DOI]
[8] Li S, Meng W, Guan Z et al. The hypoxia-related signaling pathways of vasculogenic mimicry in tumor treatment. Biomed Pharmacother, 2016; 80: 127-135.[DOI]
[9] Wei X, Chen Y, Jiang X et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol Cancer, 2021; 20: 7.[DOI]
[10] Sun B, Zhang D, Zhao N et al. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget, 2017; 8: 30502-30510.[DOI]
[11] Lu X, Sun W, Ge C et al. Contribution of the PI3K/MMPs/Ln-5γ2 and EphA2/FAK/Paxillin signaling pathways to tumor growth and vasculogenic mimicry of gallbladder carcinomas. Int J Oncol, 2013; 42: 2103-2115.[DOI]
[12] Wang M, Zhao X, Zhu D et al. HIF-1α promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up-regulation in hypoxic tumor microenvironment. J Exp Clin Cancer Res, 2017; 36: 60.[DOI]
[13] Xu Y, Li Q, Li X et al. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J Exp Clin Cancer Res, 2012; 31: 16.[DOI]
[14] Viallard C, Larrivée B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis, 2017; 20: 409-426.[DOI]
[15] Wang K, Chen Q, Liu N et al. Recent advances in, and challenges of, anti-angiogenesis agents for tumor chemotherapy based on vascular normalization. Drug Discov Today, 2021; 26: 2743-2753.[DOI]
[16] Hendrix MJC, Seftor EA, Seftor REB et al. Tumor cell vascular mimicry: Novel targeting opportunity in melanoma. Pharmacol Ther, 2016; 159: 83-92.[DOI]
[17] Ma CO, Martínez-Poveda B, Quesada AR et al. Metabolism within the tumor microenvironment and its implication on cancer progression: An ongoing therapeutic target. Med Res Rev, 2019; 39: 70-113.[DOI]
[18] Sanctis FD, Ugel S, Facciponte J et al. The dark side of tumor-associated endothelial cells. Semin Immunol, 2018; 35: 35-47.[DOI]
[19] Wilkus K, Brodaczewska K, Kajdasz A et al. Distinctive properties of endothelial cells from tumor and normal tissue in human breast cancer. Int J Mol Sci, 2021; 22: 8862.[DOI]
[20] Bani M, Decio A, Giavazzi R et al. Contribution of tumor endothelial cells to drug resistance: Anti-angiogenic tyrosine kinase inhibitors act as p-glycoprotein antagonists. Angiogenesis, 2017; 20: 233-241.[DOI]
[21] Testa U, Pelosi E, Castelli G. Endothelial progenitors in the tumor microenvironment. Adv Exp Med Biol, 2020; 1263: 85-115.[DOI]
[22] Hida K, Maishi N, Yu S et al. Heterogeneity of tumor endothelial cells and drug delivery. Adv Drug Deliv Rev, 2016; 99: 140-147.[DOI]
[23] Palikuqi B, Nguyen DT, Li G et al. Adaptable haemodynamic endothelial cells for organogenesis and tumorigenesis. Nature, 2020; 585: 426-432.[DOI]
[24] Ohga N, Ishikawa S, Maishi N et al. Heterogeneity of tumor endothelial cells: Comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am J Pathol, 2012; 180: 1294-1307.[DOI]
[25] Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer, 2013; 13: 871-882.[DOI]
[26] Gopinath P, Natarajan A, Sathyanarayanan A et al. The multifaceted role of matricellular proteins in health and cancer, as biomarkers and therapeutic targets. Gene, 2022; 815: 146137.[DOI]
[27] Tirpe AA, Gulei D, Ciortea SM et al. Hypoxia: Overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int J Mol Sci, 2019; 20: 6140.[DOI]
[28] Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol Life Sci, 2020; 77: 1745-1770.[DOI]
[29] Engel C, Brügmann G, Lambing S et al. RIG-I resists hypoxia-induced immunosuppression and dedifferentiation. Cancer Immunol Res, 2017; 5: 455-467.[DOI]
[30] Mahase S, Rattenni RN, Wesseling P et al. Hypoxia-mediated mechanisms associated with antiangiogenic treatment resistance in glioblastomas. Am J Pathol, 2017; 187: 940.[DOI]
[31] Kuczynski EA, Vermeulen PB, Pezzella F et al. Vessel co-option in cancer. Nat Rev Clin Oncol, 2019; 16: 469-493.[DOI]
[32] Stacker SA, Achen MG. The VEGF signaling pathway in cancer: The road ahead. Chin J Cancer, 2013; 32: 297-302.[DOI]
[33] Qian C, Tan M, Yang J et al. Revisiting tumor angiogenesis: Vessel co-option, vessel remodeling, and cancer cell-derived vasculature formation. Chin J Cancer, 2016; 35: 10.[DOI]
[34] Haas G, Fan S, Ghadimi M et al. Different forms of tumor vascularization and their clinical implications focusing on vessel co-option in colorectal cancer liver metastases. Front Cell Dev Biol, 2021; 9: 612774.[DOI]
[35] Motiejūnaitė R, Aranda J, Kazlauskas A. Pericytes prevent regression of endothelial cell tubes by accelerating metabolism of lysophosphatidic acid. Microvasc Res, 2014; 93: 62-71.[DOI]
[36] Weisshardt P, Trarbach T, Dürig J et al. Tumor vessel stabilization and remodeling by anti-angiogenic therapy with bevacizumab. Histochem Cell Biol, 2012; 137: 391-401.[DOI]
[37] Arjaans M, Oude Munnink TH, Oosting SF et al. Bevacizumab-induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res, 2013; 73: 3347-3355.[DOI]
[38] Bentolila LA, Prakash R, Mihic-Probst D et al. Imaging of angiotropism/vascular co-option in a murine model of brain melanoma: Implications for melanoma progression along extravascular pathways. Sci Rep, 2016; 6: 23834.[DOI]
[39] Farnsworth RH, Lackmann M, Achen MG et al. Vascular remodeling in cancer. Oncogene, 2014; 33: 3496-3505.[DOI]
[40] Jeong HS, Jones D, Liao S et al. Investigation of the lack of angiogenesis in the formation of lymph node metastases. J Natl Cancer Inst, 2015; 107: 155.[DOI]
[41] Yang J, Liao Y, Mai D et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: A meta-analysis. Angiogenesis, 2016; 19: 191-200.[DOI]
[42] Kruizinga RC, Jonge HJM, Kampen KR et al. Vascular endothelial growth factor A isoform mRNA expression in pediatric acute myeloid leukemia. Pediatr Blood Cancer, 2011; 56: 294-297.[DOI]
[43] Melincovici CS, Boşca AB, Şuşman S et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol, 2018; 59: 455-467.
[44] Hamerlik P, Lathia JD, Rasmussen R et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med, 2012; 209: 507-520.[DOI]
[45] Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol, 2016; 17: 611-625.[DOI]
[46] Zhou W, Liu K, Zeng L et al. Targeting VEGF-A/VEGFR2 Y949 signaling-mediated vascular permeability alleviates hypoxic pulmonary hypertension. Circulation, 2022; 146: 1855-1881.[DOI]
[47] Pedrosa AR, Bodrug N, Gomez-Escudero J et al. Tumor angiogenesis is differentially regulated by phosphorylation of endothelial cell focal adhesion kinase tyrosines-397 and -861. Cancer Res, 2019; 79: 4371-4386.[DOI]
[48] Li X, Padhan N, Sjöström EO et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat Commun, 2016; 7: 11017.[DOI]
[49] Tornavaca O, Chia M, Dufton N et al. ZO-1 controls endothelial adherens junctions, cell–cell tension, angiogenesis, and barrier formation. J Cell Biol, 2015; 208: 821-838.[DOI]
[50] Ju R, Zhuang Z, Zhang J et al. Angiopoietin-2 secretion by endothelial cell exosomes: Regulation by the phosphatidylinositol 3-kinase (PI3K)/Akt/endothelial nitric oxide synthase (eNOS) and syndecan-4/syntenin pathways. J Biol Chem, 2014; 289: 510.[DOI]
[51] Lee MY, Luciano AK, Ackah E et al. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci USA, 2014; 111: 12865-12870.[DOI]
[52] Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol, 2018; 9: 978.[DOI]
[53] Bertozzi D, Iurlaro R, Sordet O et al. Characterization of novel antisense HIF-1α transcripts in human cancers. Cell Cycle, 2011; 10: 3189-3197.[DOI]
[54] Jes-Niels B, Nicolas J, Heumüller AW et al. Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res, 2015; 117: 884-890.[DOI]
[55] Zhao L, Ma R, Zhang L et al. Inhibition of HIF-1a-mediated TLR4 activation decreases apoptosis and promotes angiogenesis of placental microvascular endothelial cells during severe pre-eclampsia pathogenesis. Placenta, 2019; 83: 8-16.[DOI]
[56] Skuli N, Liu L, Runge A et al. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood, 2009; 114: 469-477.[DOI]
[57] Yu B, Miao ZH, Jiang Y et al. c-Jun protects hypoxia-inducible factor-1alpha from degradation via its oxygen-dependent degradation domain in a nontranscriptional manner. Cancer Res, 2009; 69: 7704-7712.[DOI]
[58] Yu B, Li MH, Wang W et al. Pseudolaric acid B-driven phosphorylation of c-Jun impairs its role in stabilizing HIF-1alpha: a novel function-converter model. J Mol Med (Berl), 2012; 90: 971-981.[DOI]
[59] Zhang J, Qiao L, Liang N et al. Vasculogenic mimicry and tumor metastasis. J Buon, 2016; 21: 533.
[60] Delgadobellido D, Serranosaenz S, Fernándezcortés M et al. Vasculogenic mimicry signaling revisited: Focus on non-vascular VE-cadherin. Mol Cancer, 2017; 16: 65.[DOI]
[61] Thieblemont C, Molina T, Davi F. Optimizing therapy for nodal marginal zone lymphoma. Blood, 2016; 127: 2064-2071.[DOI]
[62] Kim IS, Heilmann S, Kansler ER et al. Microenvironment-derived factors driving metastatic plasticity in melanoma. Nat Commun, 2017; 8: 14343.[DOI]
[63] Strizzi L, Sandomenico A, Margaryan NV et al. Effects of a novel Nodal-targeting monoclonal antibody in melanoma. Oncotarget, 2015; 6: 34071-34086.[DOI]
[64] Benjakul N, Prakobphol N, Tangshewinsirikul C et al. Notch signaling regulates vasculogenic mimicry and promotes cell morphogenesis and the epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma. PLos One, 2022; 17: e0279001.[DOI]
[65] Tewari KS, Sill MW, Penson RT et al. Bevacizumab for advanced cervical cancer: final overall survival and adverse event analysis of a randomised, controlled, open-label, phase 3 trial (Gynecologic Oncology Group 240). Lancet, 2017; 390: 1654-1663.[DOI]
[66] Sugawara S, Lee JS, Kang JH et al. Nivolumab with carboplatin, paclitaxel, and bevacizumab for first-line treatment of advanced nonsquamous non-small-cell lung cancer. Ann Oncol, 2021; 32: 1137-1147.[DOI]
[67] Grabowski J, Glode A. Ramucirumab: A vascular endothelial growth factor receptor-2 inhibitor with activity in several malignancies. Am J Health Syst Pharm, 2016; 73: 957.[DOI]
[68] Lamdan H, Ayala M, Rojas G et al. Isolation of a novel neutralizing antibody fragment against human vascular endothelial growth factor from a phage-displayed human antibody repertoire using an epitope disturbing strategy. J Biotechnol, 2011; 151: 166-174.[DOI]
[69] Ebrahimizadeh W, Javidan Z, Rajabibazl M. Production of novel VHH nanobody inhibiting angiogenesis by targeting binding site of VEGF. Appl Biochem Biotechnol, 2015; 176: 1985-1995.[DOI]
[70] Raddum AM, Hollås H, Shumilin IA et al. The native structure of annexin A2 peptides in hydrophilic environment determines their anti-angiogenic effects. Biochem Pharmacol, 2015; 95: 1-15.[DOI]
[71] Tan H, Yang S, Liu C et al. Enhanced anti-angiogenesis and anti-tumor activity of endostatin by chemical modification with polyethylene glycol and low molecular weight heparin. Biomed Pharmacother, 2012; 66: 648-654.[DOI]
[72] Kim JH, Park I, Lee JL. Pazopanib versus sunitinib for the treatment of metastatic renal cell carcinoma patients with poor-risk features. Cancer Chemother Pharmacol, 2016; 78: 1-8.[DOI]
[73] Roskoski RJr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol Res, 2021; 165: 105463.[DOI]
[74] Kong Y, Sun L, Hou Z et al. Apatinib is effective for treatment of advanced hepatocellular carcinoma. Oncotarget, 2017; 8: 105596-105605.[DOI]
[75] Li S. Anlotinib: A novel targeted drug for bone and soft tissue sarcoma. Front Oncol, 2021; 11: 664853.[DOI]
[76] Parekh H, Griswold J, Rini B. Axitinib for the treatment of metastatic renal cell carcinoma. Future Oncol, 2016; 12: 303-311.[DOI]
[77] Dadashpour S, Küçükkılınç TT, Ercan A et al. Synthesis and anticancer activity of benzimidazole/benzoxazole substituted triazolotriazines in hepatocellular carcinoma. Anticancer Agents Med Chem, 2019; 19: 2120-2129.[DOI]
[78] Cai W, Ratnayake R, Wang M et al. Inhibition of cotranslational translocation by apratoxin S4: Effects on oncogenic receptor tyrosine kinases and the fate of transmembrane proteins produced in the cytoplasm. Curr Res Pharmacol Drug Discov, 2021; 2: 100053.[DOI]
[79] Xie S, Ogden A, Aneja R et al. Microtubule-Binding Proteins as Promising Biomarkers of Paclitaxel Sensitivity in Cancer Chemotherapy. Med Res Rev, 2016; 36: 300-312.[DOI]
[80] Zhang Y, Gong F, Lu Z et al. DHPAC, a novel synthetic microtubule destabilizing agent, possess high anti-tumor activity in vincristine-resistant oral epidermoid carcinoma in vitro and in vivo. Int J Biochem Cell Biol, 2017; 93:1-11.[DOI]
[81] Pérezpérez MJ, Priego EM, Bueno O et al. Blocking blood flow to solid tumors by destabilizing tubulin: an approach to targeting tumor growth. J Med Chem, 2016; 59: 8685-8711.[DOI]
[82] Gong F, Wang L, Yu L et al. DHPAC, a novel microtubule depolymerizing agent, suppresses angiogenesis and vasculogenic mimicry formation of human non-small cell lung cancer. J Cell Biochem, 2020; 121: 4756-4771.[DOI]
[83] Xu L, Wang W, Meng T et al. New microtubulin inhibitor MT189 suppresses angiogenesis via the JNK-VEGF/VEGFR2 signaling axis. Cancer Lett, 2018; 416: 57-65.[DOI]
[84] Rakesh KJ. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell, 2014; 26: 605-622.[DOI]
[85] Wong PP, Demircioglu F, Ghazaly E et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell, 2015; 27: 123-137.[DOI]
[86] Fukumura D, Kloepper J, Amoozgar Z et al. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol, 2018; 15: 325-340.[DOI]
[87] Huang Y, Kim BYS, Chan CK et al. Improving immune–vascular crosstalk for cancer immunotherapy. Nat Rev Immunol, 2018; 18: 195-203.[DOI]
Copyright © 2023 The Author(s). This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.


