Virtual Screening, Molecular Dynamics, MM-PBSA and In-silico Studies in Search of Potent Novel Fibroblast Growth Factor Receptors 4 Inhibitor
Fady H Fayek1*, Omar M Rashad2
1Department of Pharmaceutical Chemistry, Faculty of Pharmacy and Drug Technology, Egyptian Chinese University, Cairo, Egypt
2Department of Pharmaceutics, Faculty of Pharmacy, Galala University, Suez, Egypt
*Correspondence to: Fady H Fayek, MSc, Department of Pharmaceutical Chemistry, Faculty of Pharmacy and Drug Technology, Egyptian Chinese University, Gesr ElSuez st, Cairo, 11786, Egypt; Email: Habib.Fady@yahoo.com
Abstract
Objective: The objective of this study was to design a virtual screening (VS) protocol for identifying potential fibroblast growth factor receptor 4 (FGFR4) inhibitors from the commercially available Specs database. This was based on an experimental model of FGFR4 bound to the potent inhibitor FGF401 (PDB: 7VJL), with the aim of discovering novel compounds with therapeutic potential for various tumor types.
Methods: The study comprised two main parts. Firstly, a ligand-based pharmacophore model was constructed using known potent FGFR4 inhibitors to discriminate between actives and decoys. Secondly, docking studies of the identified actives were conducted within the binding pocket of the FGFR4 experimental model. This aimed to assess the ability of the compounds to overlay on the reference inhibitor and interact similarly within the binding site. The virtual screening protocol was then applied to the Specs database to identify potential FGFR4 inhibitors.
Results: The ligand-based pharmacophore model exhibited promising discrimination capabilities, achieving a true positive percentage of 100% and a false positive percentage of 7%. Docking studies revealed that the selected compounds overlaid well with the reference inhibitor and exhibited interactions within the FGFR4 binding pocket. Through the virtual screening of the Specs database, compound A emerged as the most suitable candidate. Molecular dynamics simulation and Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) calculations further supported the efficacy of compound A as a FGFR4 inhibitor. Additionally, compound A demonstrated favorable absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, along with adherence to Lipinski’s rule of 5, enhancing its potential as a lead compound.
Conclusion: In conclusion, compound A emerged as a promising selective FGFR4 inhibitor with potential for therapeutic use in various cancers. Further in vitro experiments and animal cancer model studies are warranted to validate its efficacy. The study highlights the effectiveness of the proposed virtual screening protocol in identifying novel FGFR4 inhibitors, offering a promising avenue for future drug discovery efforts in oncology.
Keywords: FGFR4, fibroblast growth factor receptor 4, FGF401, virtual screening, docking, pharmacophore, molecular dynamics, in-silico studies, cancer
1 INTRODUCTION
Fibroblast growth factor receptors (FGFRs) are a family of receptor tyrosine kinases (RTKs) that play a crucial role in various cellular processes such as cell proliferation, differentiation, migration, and survival. Studies have shown that, the differential expression of the ligand fibroblast growth factors (FGFs) and their receptors throughout the developing and mature gastrointestinal tract that seem to play an important role in modulating gut development and epithelial repair mechanisms after injury[1-3].
The FGFR family is composed of four members FGFR1-4, which are activated by binding to fibroblast growth factors. Upon ligand binding, FGFRs undergo dimerization and autophosphorylation leading to the activation of downstream signaling pathways such as the MAPK and PI3K/Akt pathways. In humans, dysregulation of the FGFR signaling is associated with a variety of diseases such as cancers, skeletal dysplasia and developmental disorders. As a result, FGFRs have become an attractive therapeutic target for the treatment of these diseases[4-6].
Recent studies have shown a clear understanding of FGFRs structure consisting of an extracellular ligand binding domain, a single-transmembrane domain, and a split intracellular kinase domain, also their function and regulation, as well as their role in the development of disease. For instance, mutations in FGFRs have been identified in several types of cancer, including breast, lung, and bladder cancer, and have been shown to drive tumor growth and progression. In addition, FGFR inhibitors have shown promising results in the preclinical and clinical studies for the treatment of cancer and other disorders. But due to the complexity of FGFR signals and potential for off target effect, it remains a challenge to develop an effective inhibitor of FGFR. Hence, more research is needed to understand how FGFR signals work and develop inhibitors with greater efficiency and selectivity for clinical use[4-7].
The fibroblast growth factor receptor 4 (FGFR4) is a transmembrane receptor protein that plays a critical role in various cellular processes, including cell growth, differentiation, and survival. Dysregulation of FGFR4 has been implicated in the development and progression of several types of cancer, including breast, liver, lung, and prostate cancer[8,9].
The activation of downstream signaling pathways like PI3K/Akt as well as MAPK/ERK, which are involved in the promotion of cell proliferation and survival is an important mechanism by which FGFR4 contributes to cancer. By interacting with extracellular matrix proteins, FGFR4 has also been shown to promote the invasion of cancer cells and metastasis[5,10-13].
In breast cancer, high levels of FGFR4 expression have been associated with poor prognosis and resistance to chemotherapy. In liver cancer, FGFR4 has been shown to play a critical role in the development of hepatocellular carcinoma (HCC), and FGFR4 inhibitors have shown promise as a potential therapeutic option for HCC. FGFR4 has been linked to the development of squamous cell carcinoma in lung cancer, and an inhibitor of FGFR4 is found to have a beneficial effect on preclinical models. Moreover, FGFR4 has been shown to promote survival of tumor cells and resistance to androgen deprivation therapy in prostate cancer[5,10].
Several studies have also reported genetic variations in FGFR4 that are associated with increased cancer risk. The risk of breast, prostate or lung tumors is increased by polymorphisms in the FGFR4 gene that are referred to as a G388R polymorphism. Overall, these data indicate that FGFR4 represents a promising target for the development of novel cancer therapies. Therefore, there are several FGFR4 inhibitors being developed and results have been promising in a number of different types of cancer[5,8,10,14].
FGF401 is a potent and selective inhibitor of FGFR4 and has minimal activity against other FGFR family members, this high selectivity for FGFR4 is important because it reduces the risk of off-target effects and potential toxicity. In preclinical studies, FGF401 has been shown to bind within an ATP binding pocket of FGFR4, inhibiting its kinase activity and triggering inhibition of downstream signal transduction pathways critical for the growth and survival of cancer cells. Thus, it has shown promise as a potential therapeutic option for patients with HCC, a type of liver cancer that is often resistant to standard chemotherapy and radiation therapy[10,15-17].
FGF401 was shown to have promising safety and preliminary efficacy results in Phase 1 clinical trial in patients with advanced HCC or other solid tumors by its inhibitory effect to FGFR4. The studies have shown that FGF401 is well-tolerated and has a moderate safety profile, with most of undesirable effects occurring due to fatigue, nausea or diarrhea. In terms of efficacy, FGF401 showed promising antitumor activity, with partial responses observed in some patients with HCC and other solid tumors. Following these outcomes, a Phase 2 clinical trial with FGF401 is under way in patients with HCC[10,15].
Finally, further clinical studies are needed to determine the safety and efficacy of FGF401 in larger patient populations and to assess its potential as a targeted therapy for FGFR4-driven cancers.
2 METHODOLOGY
2.1 Pharmacophore
2.1.1 Compounds Preparation
Previously reported active compounds having affinity to FGFR4 with IC50≤100nM were collected in one database entitled with FGFR4 inhibitors[3,9,18,19]. These compounds were prepared using Molecular Operating Environment (MOE) version 2019.01 software through three steps which are washing, partial charge calculation and energy minimization using force field MMFF94x with the default gradient 0.1 RMS kcal/(mol.Å).
2.1.2 Flexible Alignment of FGFR4 Inhibitors
The low energy conformers of 19 compounds previously reported as FGF4 protein inhibitors were aligned using flexible alignment tool in MOE version 2019.01[20]. A stochastic conformational search with an energy window of 15kcal/mol was conducted to compute a collection of alignments with an iteration limit of 1,000 and a failure limit of 50. The resultant alignments were evaluated based on F, S and U scores. The F score detects the similarity of each configuration. The U score is the average strain energy of the molecules in the alignment in kcal/mol. The S score, also called the grand alignment score, is obtained by summation of the U and F scores.
2.1.3 Pharmacophore Model Generation
The pharmacophore model was developed by using the pharmacophore features and projected pharmacophore features based on the Unified annotation scheme the pharmacophore consensus tool of the pharmacophore query editor in MOE. The shortest allowed distance between features was set to 1.3Å with the generated features matching 85% of the active compounds. The generated pharmacophore model was validated by using a test set database composed of multi conformational 19 actives compounds collected from past literature and 1,100 decoys generated by Directory of Useful Decoys (DUD-E) website[21,22]. The test set compounds were mapped to the pharmacophore model using the pharmacophore search tool available in MOE. The performance of the pharmacophore model was assessed based on the number of active and inactive compounds matching the pharmacophore model where the true positive (TP%) and false positive (FP%) percentages were calculated.
2.2 Molecular Docking
MOE version 2019.10 software was employed for molecular docking using an experimental X-ray FGFR4 protein bound to the FGFR4 inhibitor FGF401 (PDB:7VJL). This protein was prepared for docking with Quick prep wizard found in MOE[20].
The binding pocket residues of FGFR4 protein were defined based on the previously reported binding mode of FGF401 (a potent FGFR4 inhibitor)[3,9,18]. In order to assess the reliability and robustness of our docking algorithm, the minimized form of FGF401 was first docked into the binding pocket of FGFR4 protein using different replacement, refinement methods, scoring functions in order to reach the optimum docking algorithm. The resultant conformations were evaluated by the score of docking (S), the root mean square deviation (RMSD) with respect to the co-crystallized ligand FGF401 and fulfilling the crucial interactions with the important reported amino acid residues in the binding pocket of the receptor. The docking protocol in the FGFR4 binding site was further assessed in terms of enrichment performance by retrospective screening of a validation test set database of 382 compounds. A set of 18 previously reported FGFR4 inhibitors along with 364 decoys generated through DUD-E website were docked into FGFR4.
2.3 Prospective Virtual Screening
A total of 538,920 commercially available compounds were downloaded from Specs website[23]. The validated pharmacophore model was utilized as a 3D query for screening the commercially available Specs database. Compounds were prepared as described in the compound preparation section, filtered using molecular weight (MW) descriptor to identify compounds within the same weight range of the active compounds (356:636Da). The prepared compounds were saved in smi file format and 3D conformations were generated using the online Pharmit server[24]. This server uses a knowledge-based search protocol to generate 3D conformations. The resultant conformations were mapped to the pharmacophore model such that hit molecules match all of the query features. Using MOE, the identified hit compounds were docked into the binding pocket of FGFR4.
2.4 Molecular Dynamics (MD) Simulation
MD simulations enable a better understanding of ligand through a number of statistical characteristics, which are used to analyze the stability of ligand in the protein binding site[25,26]. The best docked pose of compound A along with FGFR4 complex was chosen as starting coordinates for 100ns MD simulation using a GROMACS-2023 software package (GNU, General Public License[27] and CHARMM36 force field[28,29].The complex of compound A and FGFR4 was solvated within a cubic box of dimensions (100×100×100Å) using transferable intermolecular potential with a three-points (TIP3P) water model, thus allowed a margin of 10Å minimum between the protein and each side of the 3D box. The complex was also prepared for the simulation through ionization and neutralization steps at physiological pH7.0, via the addition of Na+ and Cl- ions using Monte-Carlo ion-placing method[30]. The MD simulation was performed in three stages (minimization, equilibration, and production), the minimization and equilibration stages were conducted using a 1000kJ/mol·nm2 force constant to constrain all heavy atoms and maintain the original protein folding[30]. The minimization step comprised the initial optimization of each system's shape using the steepest descent algorithm over 5,000 iterations (5ps). The equilibration step involved system equilibration for 125ps under constant number of particles, volume, and temperature ensemble (NVT ensemble), followed by 125ps under constant pressure ensemble (NPT ensemble), both of which were guided by the Parrinello-Rahman barostat and the Berendsen temperature coupling method, respectively, to control the temperature within 3D box[31,32]. Finally, the production stage was run for 100ns under NPT ensemble, in which, the isothermic and isobarometric conditions were maintained at 300K temperature and 1atm pressure using the Parrinello-Rahman barostat. the Particle Mesh Ewalds method was used to calculate the long-range electrostatic interactions[33]. The time step of the simulated system was set to 2.0fs. The trajectories generated from the simulation were stored every 100ps. These trajectories were used to compute RMSD, root mean square fluctuation (RMSF), solvent accessible surface area (SASA), radius of gyration, and number of hydrogen bonds. Plots and visual inspection of the trajectories was done using XMgrace[34] and VMD[35], respectively.
2.5 MMPBSA Calculations
The binding free energy in our work was calculated using the MM/PBSA[36] technique through gmx_mmpbsa tool[37] of GROMACS 2023 package. The following formulas are used to compute the binding free energy (ΔGbinding) in MM-PBSA between a ligand and a receptor to create a complex:
ΔGbinding=Gcomplex-(Greceptor+Gligand) (1)
ΔGbinding=ΔEMM+ΔGsolvation-TΔS (2)
EMM=ΔEELE+ΔEVDW (3)
ΔGsolvation=ΔGnonpolar/sol+ΔGpolar/sol (4)
where the system's binding free energy is represented by ΔGbinding, the overall energy of the protein-ligand complex is indicated by the Gcomplex, and the free energies of the protein and ligand in solvents are indicated by the receptor and ligand, respectively. Van der Waals (ΔEVDW) and electrostatic (ΔEELE) energies are also included in the representation of the gas phase interaction energy, which is denoted by ΔEMM. The polar and nonpolar desolvation energy contributions were ascertained using SASA and PB, respectively. The polar and nonpolar components of the desolvation free energy[38] are represented by ΔGsolvation. TΔS (this term is ignored) represents the change in conformational entropy during ligand binding[39], where T is the absolute temperature and ΔS is the entropy change. However, in a real application, it is more common to simply mimic the complex state that leads to the cancellation of ΔEINT. As a result, the binding free energy calculation equation is changed to:
ΔGbinding=ΔEMM+ΔGsolvation-TΔS=ΔEVDW+ΔGpolar/sol+ΔGnonpolar/sol-TΔS (5)
2.6 In-Silico IC50 Prediction
Half-maximal inhibitory concentration value (IC50 value) is considered a tool of measuring the efficacy of a given compound in inhibiting a desired target. Acceptable IC50 value is needed for proceeding the upcoming steps in drug development. IC50 values of the best docking score specs database compound and the reference FGFR4 inhibitor, FGF401 were calculated using Cell-Line Cytotoxicity Predictor[40]. It is a dedicated tool based on experimental data for evaluation of cytotoxicity against cancer cell lines. The compounds were introduced to the website in SMILES format and the Pa value was set to 0.3.
2.7 ADMET Properties Prediction
The study of absorption, distribution, metabolism, and excretion (ADME) properties is crucial to interpret the characteristics of the drug being developed inside our bodies. In addition to ADME properties, also studying the toxicity (T) profile also deserves thoroughly importance because in some cases intermediate metabolites can show toxicity to the body[41]. Drug development is a long, costly, and tiresome process. Early detection of unsuitable absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties can save money, time, and effort. In silico tools can be implemented in some situations as an alternative to animal testing in predicting these properties[11,42-44]. ADMET properties of the first ranked compound in Specs database docking (compound A) and the reference compound FGF401 were predicted and compared using the well-known online tool pkCSM[45].
3 RESULTS AND DISCUSSION
3.1 Pharmacophore
3.1.1 Alignment and Pharmacophore Generation
The pharmacophore features were generated by aligning low-energy conformers of the 19 previously reported active compounds. Structural alignment of the actives resulted in 20 different configurations. The alignment with best U score was chosen to be used as a basis to search for the common pharmacophore features of the FGFR4 inhibitors (Figure 1).
|
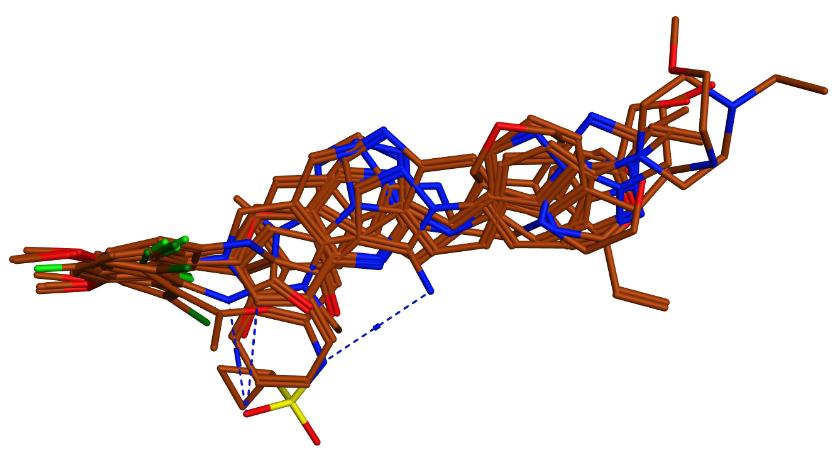
Figure 1. Structural alignment of the FGFR4 inhibitor.
The pharmacophore model was generated using the pharmacophore consensus tool of the pharmacophore query editor in MOE. The model was generated by setting the shortest allowed distance between features to 1.3Å, with the generated features matching 85% of the active compounds. The generated model comprised 5 features which are 2 hydrophobic/aromatic centers (Hyd/Aro, 1Å), 1 pi electron feature (PiN, 1.1Å) and 2 hydrogen bond acceptor feature (Acc, 0.6Å and 1Å) (Figure 2). This appears in agreement with previous literature where the co-crystallized FGF401 was reported to form hydrophobic interactions with Leu493, Ala554 of FGFR4 protein, and also hydrogen bond interactions with Lys503 and Ala553 residues[46] (Figure 3). Pharmacophore model validation was done by screening the multi-conformation test set compounds which includes 19 FGFR4 inhibitors and 1,100 compounds with similar physical properties as actives MW, number of hydrogen-bond donors and hydrogen-bond acceptors, number of rotatable bonds, and octanol-water partition coefficient (logP), labeled as decoys. The performance of the pharmacophore model was assessed based on the number of actives and inactives matching the pharmacophore model by calculating the true positive (TP%) and false positive (FP%) percentages. All of the 19 active compounds appeared matching the pharmacophore model with a TP% of 100%. On the other hand, only 77 compounds out of a total of 1,100 inactive compounds matched the pharmacophore model yielding a 7 FP%.
|
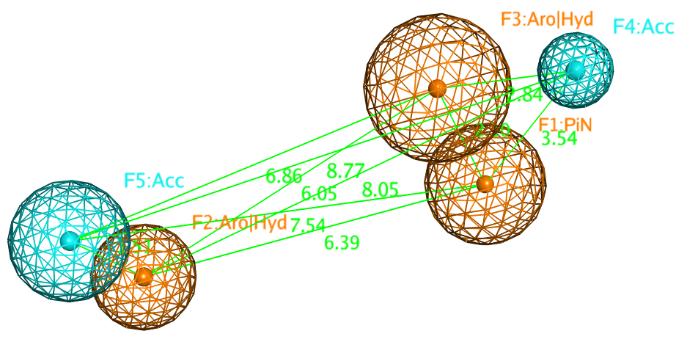
Figure 2. Distance constraints between the chemical features of the pharmacophore model represented by orange (pi electron, aromatic centers and its’ projections), and cyan (hydrogen bond acceptor) spheres.
|
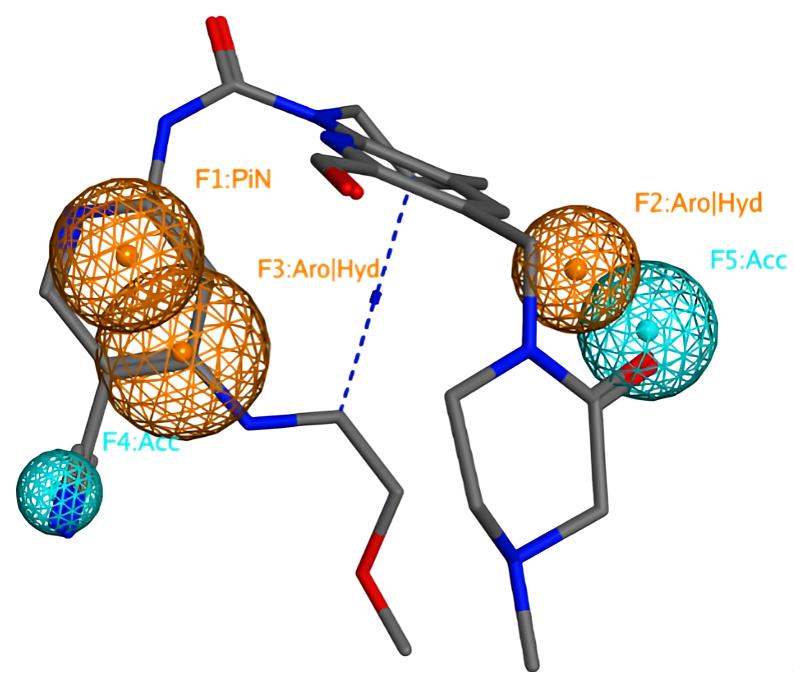
Figure 3. The reference compound mapped onto the pharmacophore model. The unified annotation scheme included in MOE (2019.01) was used.
3.2 Molecular Docking
Molecular docking experiments were performed using MOE 2019.10. A previously published X-ray crystallography model of the human FGFR4 protein bound to the potent inhibitor FGF401 (PDB:7VJL) was used in this study. FGF401 was first docked into the binding pocket of the FGFR4 protein to examine the reliability of the binding modes produced. All the resultant poses converged to a binding mode similar to the co-crystallized ligand with the best ranking pose having a RMSD value of 0.6Å. The scoring function used was alpha hydrogen bond due to it’s confident performance in discrimination between actives and inactive compounds, and also rank the correct poses over the others, the placement method utilized to predict the right binding poses was triangle matcher with induced fit refinement. The produced binding poses were similar to the reported binding pose of FGFR4, which ensures the reliability of our algorithm (Figure 4). The performance of the chosen docking algorithm was further evaluated for actives enrichment by docking a test set comprising 18 FGFR4 inhibitors and 364 decoys generated through DUD-E website[22], and the enrichment factor at 5% (EF5%) was 5.87.
|
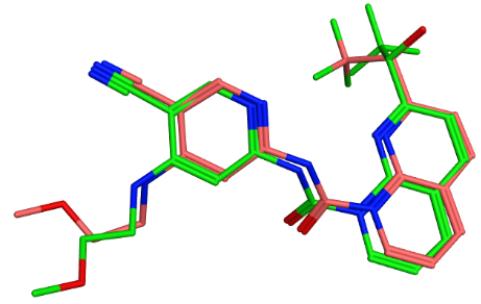
Figure 4. The overlaying of the co-crystallized ligand FGF401 (pink color) and the best docked pose of FGF401 (green).
3.3 Prospective Virtual Screening
The previously described validated methods were used to find new hit compounds that can bind to the binding site of FGFR4. A total of 538,920 compounds were downloaded from the SPECS website[7]. These compounds were initially screened using MW descriptor to identify compounds within the same weight range of the active compounds (356:636Da). A total of 25,919 compounds were in the same weight range of the previously reported FGFR4 inhibitors. These filtered hits were screened using the validated pharmacophore model in order to exclude the compounds lacking the essential chemical features for binding to FGFR4. A total of 7,556 compounds matched our pharmacophore model representing 5,257 different chemical scaffolds. To examine their binding modes, these hit compounds were docked into the binding pocket of FGFR4 protein using MOE 2019[20]. The top 1% ranked compounds from the previous mentioned docking procedure were selected for visual inspection. The resultant compounds were clustered using the scaffold classification script in MOE. Upon visual examination of the binding modes of the different cluster representatives, the 1st ranked docked compound (compound A) displayed a promising binding mode towards the protein binding site with nearly the same critical interactions as the co-crystallized ligand FGF401, in addition to one more hydrogen bond interaction with Asp630 and hydrophobic interaction with Gly479 due to the extended structure of our compound compared to the co-crystallized ligand (Figure 5). The docking score for compound A was -52.919 while that of FGF401 was -48.565, which indicates that compound A has a superior fit in the binding pocket compared to FGF401.
|

Figure 5. The super-imposition of compound A (green) and FGF401 (red), revealing their interactions with the protein binding site.
3.4 MD Simulation
MD simulation was conducted to evaluate the stability of compound A and FGFR4 complex. The RMSD of the ligand inside the binding pocket of FGFR4 was calculated to assess its stability across the simulation trajectories. Compound A was found with an average of 0.208nm, which indicates its stability with least deviation within the binding pocket of FGFR4. The radius of gyration (RoG) for the complex was in the range of 1.96-2.05nm, with an average 2nm, showing the compactness of the simulated complex. SASA was also calculated and it was found that the surface area accessed by solvent molecules was within an acceptable range of 142 to 159nm2, with an average of 150.192nm2. Four hydrogen bonds were found maximally between compound A and FGFR4 throughout the time interval of the simulation (100ns), which ensures the stability of our complex. Furthermore, maximum amino acid residues in the protein had an RMSF value less than 0.49nm which assures the stability and compactness of the protein. The graphical representation plots of the different statistical parameters were presented in Figures 6 and 7. All of the gathered snapshots were used to create an average conformation of the whole MD simulation, and the snapshot that showed the greatest structural resemblance to this average conformation was selected as the representative conformation. Figure 8 displays a structural comparison between the docked pose and the MD representative conformation of compound A bound to FGFR4; the figure was prepared using PYMOL[47].
|
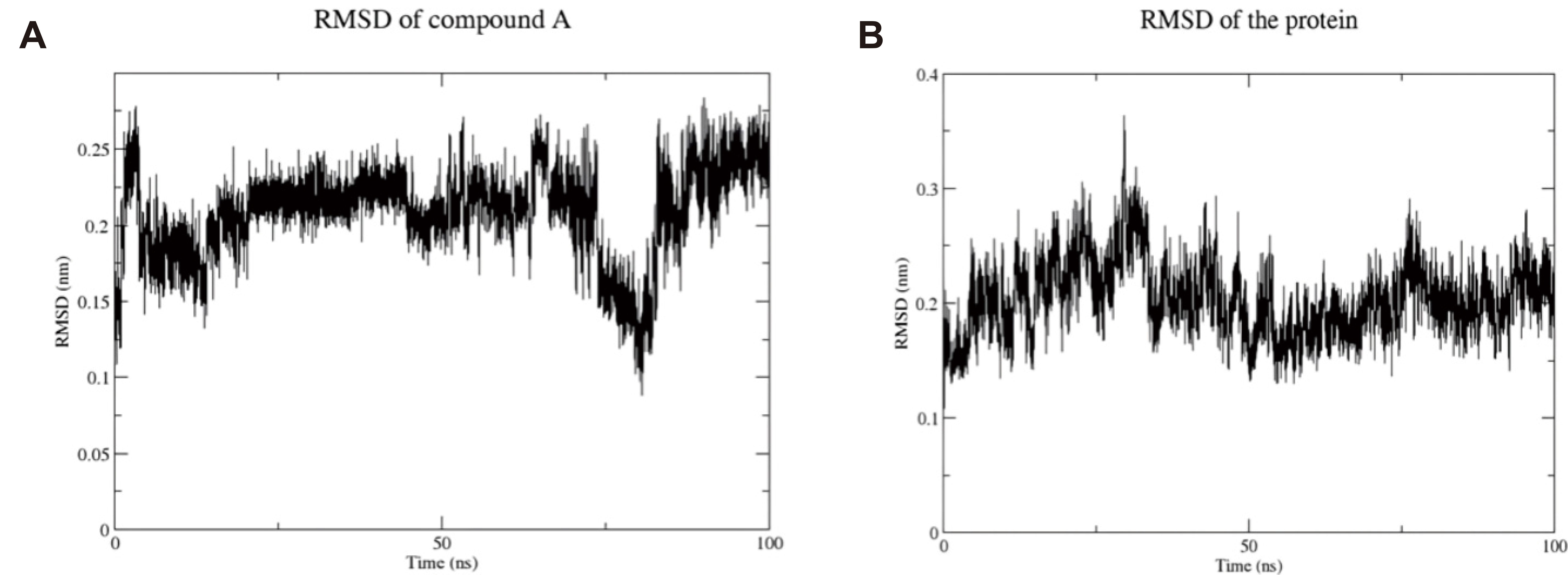
Figure 6. Root mean square deviation of compound A and FGFR4 protein throughout the whole simulation. A: Root mean square deviation of FGFR4 protein in presence of compound A; B: Root mean square deviation of compound A throughout 100ns MD simulation.
|

Figure 7. MD 100ns simulations analysis plots. A: Root mean square fluctuation of the protein amino acids residues; B: The Radius of gyration of the protein; C: Number of hydrogen bonds between compound A and FGFR4 protein; D: SASA of FGFR4.
|

Figure 8. MD representative structure of compound A (blue color) superimposed on the docked pose (Magenta color) inside the binding pocket of FGFR4.
3.5 MMPBSA Calculations
MMPBSA was used to measure the binding affinity of compound A. The binding free energy of compound A in complex with FGFR4 protein was calculated by sampling frames at a regular interval of 10ps from the whole 100ns simulation. As displayed in Table 1, CCR9-13 showed a binding free energy of -22.73kcal/mol. Compound A binding affinity to FGFR4 appeared consistent with its stability throughout the whole MD simulation.
Table 1. Binding Free Energies (in Kcal/mol) for Compound A Bound to FGFR4 Protein
Compound Name |
MM-PBSA(Kcal/mol) |
Std Deviation |
Std Error of Mean |
Compound A |
-22.73 |
2.73 |
1.33 |
3.6 In-silico IC50 Prediction
IC50 predictions showed that our suggested compound A has more probability to act on colon carcinoma (RKO cell line) than FGF401 (Table 2), in addition to this, compound A was predicted to be active on brain oligodendeoglioma, breast adenocarcinoma and non-small cell lung carcinoma with Pa values; 0.457,0.352 and 0.372 and Pi values; 0.080, 0.065 and 0.073 respectively. It is worth mentioning that Pa is the probability of belonging to the active compounds subclass, while the Pi value is the probability to be an inactive compound according to the used training set in the IC50 prediction tool.
Table 2. IC50 value Prediction
Compound |
Pa |
Pi |
Cell Line |
Compound A |
0.389 |
0.033 |
RKO (Colon carcinoma) |
FGF401 |
0.306 |
0.146 |
RKO (Colon carcinoma) |
3.7 ADMET Properties Prediction
Using the pkCSM tool, 10 toxicity parameters, 7 absorption parameters, 4 distribution parameters, 7 metabolism parameters, 2 excretion parameters, and 9 toxicity parameters for compound A and FGF401 were determined. Numerous parameters had only slight variations between the 2 compounds, but in other parameters, compound A outperformed the reference compound (Table 3). It is important to note that compound A has a high gastrointestinal tract absorption and also it demonstrated a high drug likeness; it did not violate any of the Lipinski's rule of five criteria[48] and Ghose filter[49] as well according to the online server Swiss ADME[50].
Table 3. Comparison Between Compound A and FGF401 in Terms of ADMET Properties
Property |
Model Name |
Unit |
Predicted Value |
|
Absorption |
Water solubility |
Numeric (log mol/L) |
Compound A |
FGF401 |
-4.455 |
-2.94 |
|||
Caco2 permeability |
Numeric (log Papp in 10-6 cm/s) |
0.073 |
0.686 |
|
Intestinal absorption (human) |
Numeric (% Absorbed) |
62.231 |
30.713 |
|
Skin Permeability |
Numeric (log Kp) |
-2.735 |
-2.736 |
|
P-glycoprotein substrate |
Categorical (Yes/No) |
Yes |
Yes |
|
P-glycoprotein I inhibitor |
Categorical (Yes/No) |
Yes |
No |
|
P-glycoprotein II inhibitor |
Categorical (Yes/No) |
Yes |
No |
|
Distribution |
VDss (human) |
Numeric (log L/kg) |
-0.755 |
-0.038 |
Fraction unbound (human) |
Numeric (Fu) |
0 |
0.182 |
|
BBB permeability |
Numeric (log BB) |
-1.217 |
-1.606 |
|
CNS permeability |
Numeric (log PS) |
-3.283 |
-3.382 |
|
Metabolism |
CYP2D6 substrate |
Categorical (Yes/No) |
No |
No |
CYP3A4 substrate |
Categorical (Yes/No) |
Yes |
Yes |
|
CYP1A2 inhibitor |
Categorical (Yes/No) |
No |
No |
|
CYP2C19 inhibitor |
Categorical (Yes/No) |
No |
No |
|
CYP2D6 inhibitor |
Categorical (Yes/No) |
Yes |
No |
|
CYP2D6 inhibitor |
Categorical (Yes/No) |
No |
No |
|
Excretion |
Total Clearance |
Numeric (log ml/min/kg) |
0.087 |
0.617 |
Renal OCT2 substrate |
Categorical (Yes/No) |
No |
No |
|
Toxicity |
AMES toxicity |
Categorical (Yes/No) |
No |
No |
Max. tolerated dose (human) |
Numeric (log mg/kg/day) |
0.546 |
0.408 |
|
hERG I inhibitor |
Categorical (Yes/No) |
No |
No |
|
hERG II inhibitor |
Categorical (Yes/No) |
Yes |
Yes |
|
Oral Rat Acute Toxicity (LD50) |
Numeric (mol/kg) |
2.989 |
2.893 |
|
Oral Rat Chronic Toxicity (LOAEL) |
Numeric (log mg/kg_bw/day) |
1.624 |
1.888 |
|
Skin Sensitization |
Categorical (Yes/No) |
Yes |
No |
|
T.Pyriformis toxicity |
Numeric (log ug/L) |
0.285 |
0.285 |
|
Minnow toxicity |
Numeric (log mM) |
0.402 |
2.71 |
|
4 CONCLUSION
Targeting FGFRs as anti-cancer therapy is a good topic for investigation. The majority of previous studies concentrated on FGFR 1-3, however there is mounting evidence that FGFR4 plays a significant and distinctive role in oncogenesis,tumour growth, and resistance to anti-tumor therapy in a variety of cancer types. Many compounds inhibiting FGFRs were reported previously, but with lack of selectivity. Only 4 compounds showed more selectivity towards FGFR4 on the expense of the others. FGF401 (N-[5-cyano-4-(2-methoxyethylamino) pyridin-2-yl]-7-formyl-6-[(4-methyl-2-oxopiperazin-1-yl)methyl]-3, 4-dihydro-2H-1,8-naphthyridine-1-carboxamide) displayed higher selectivity towards FGFR4 with 1,000 folds’ selectivity compared to the others, in addition to this, FGF401 has gone through clinical trials. In this study, a ligand-based pharmacophore model was developed based on the previously reported FGFR4 inhibitors. A test set was used to test the discrimination ability of the selected pharmacophore model between actives and decoys. The model gave excellent performance with a TP% of 100% and FP% of only 7%. An experimental model for FGFR4 bound to the potent FGF401 (PDB: 7VJL) obtained by X-ray crystallography was used in order to find novel FGFR4 inhibitors. This FGFR4 model was used in docking process of the past reported FGFR4 inhibitors to evaluate the reliability and the reproducibility of the model in generating binding modes agrees with those reported previously. The proposed pharmacophore model and docking algorithm were used to virtually screen the commercially available database Specs. Screening of the Specs database using our validated pharmacophore model and docking algorithm showed that compound A is the most promising compound to be a lead compound. The docking experiment identified that compound A fits well in the binding pocket of FGFR4, fulfilling the reported crucial interactions like the reference ligand; hydrogen bond with Ala553 and Lys203, in addition to, 2 extra interactions were implemented by our compound; hydrogen bond with Asp630 and hydrophobic interaction with Gly479 due to the extended structure of our compound in the binding pocket compared to the reference compound. Later, MD studies and MMPBSA calculation supported this finding. Concerning ADMET properties, compound A showed superiority on FGF401 in some parameters, while other parameters were nearly the same in both, which validates our postulate that compound A could be a potent selective FGFR4 inhibitor; which raises the chance to become a promising compound that can be used to treat various diseases associated with blocking FGFR4 like brain oligodendeoglioma, breast adenocarcinoma, non-small cell lung carcinoma and colon carcinoma. Additionally, compound A obeys Lipinski rule of 5 (MW: 462.45Da, octanol–water partition coefficient (logP): -2.44, hydrogen bond donor count: 4, and hydrogen bond acceptor count: 6; according to the online server Swiss ADME, also this compound is novel with no past activity on other targets according to Reaxys software. This study’s drawback is the absence of in vitro testing, additional research on cancer cell lines, malignancy-modeling animals, and clinical experiments can demonstrate the potential of this compound.
Acknowledgements
Not applicable.
Conflicts of Interest
The authors declared no conflict of interest.
Author Contribution
All authors contributed to the manuscript and approved the final version.
Abbreviation List
ADME, Absorption, distribution, metabolism, and excretion
ADMET, Absorption, distribution, metabolism, excretion, and toxicity
DUD-E, Directory of useful decoys
FGFR4, Fibroblast growth factor receptor 4
FGFRs, Fibroblast growth factor receptors
FGFs, Fibroblast growth factors
HCC, Hepatocellular carcinoma
MD, Molecular dynamics
MMPBSA, Molecular mechanics poisson-boltzmann surface area
MOE, Molecular operating environment
MW, Molecular weight
PME, Particle mesh ewalds
RMSD, Root mean square deviation
RMSF, root mean square fluctuation
RoG, Radius of gyration
SASA, Solvent accessible surface area
VS, Virtual screening
References
[1] Böttcher RT, Niehrs C. Fibroblast growth factor signaling during early vertebrate development. Endocr Rev, 2005; 26: 63-77.[DOI]
[2] Fang B, Lai Y, Yan H et al. Design, synthesis, and biological evaluation of 1,6-naphthyridine-2-one derivatives as novel FGFR4 inhibitors for the treatment of colorectal cancer. Eur J Med Chem, 2023; 259: 115703.[DOI]
[3] Zhang Y, Yu N. Design, Synthesis and Biological Evaluation: 5-amino-1H-pyrazole-1-carbonyl derivatives as FGFR Inhibitors. Lett Drug Des Discov, 2020; 17: 1330-1341.[DOI]
[4] Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol, 2019; 16: 105-122.[DOI]
[5] Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer, 2010; 10: 116-129.[DOI]
[6] Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer, 2017; 17: 318-332.[DOI]
[7] Gabler L, Jaunecker CN, Katz S et al. Fibroblast growth factor receptor 4 promotes glioblastoma progression: A central role of integrin-mediated cell invasiveness. Acta Neuropathol Com, 2022; 10: 65.[DOI]
[8] Chen H, Shen DP, Zhang ZZ et al. Fibroblast growth factor receptor 4 protein expression and clinicopathological features in gastric cancer. World J Gastroenterol, 2015; 21: 1838.[DOI]
[9] Wei W, Li Y, Peng C et al. Design, synthesis and biological evaluation of novel diaminopyrimidine derivatives as covalent fibroblast growth factor receptor 4 inhibitors. Results Chem, 2023; 5: 100893.[DOI]
[10] Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev, 2014; 34: 280-300.[DOI]
[11] Kar S, Leszczynski J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin Drug Dis, 2020; 15: 1473-1487.[DOI]
[12] Li X, Lu W, Kharitonenkov A et al. Targeting the FGF19-FGFR4 pathway for cholestatic, metabolic, and cancerous diseases. J Intern Med, 2024; 295: 292-312.[DOI]
[13] Chen X, Huang Y, Liu H et al. Insight into the design of FGFR4 selective inhibitors in cancer therapy: Prospects and challenges. Eur J Med Chem, 2023; 263: 115947.[DOI]
[14] Ma CX, Crowder RJ, Ellis MJ. Importance of PI3-kinase pathway in response/resistance to aromatase inhibitors. Steroids, 2011; 76: 750-752.[DOI]
[15] Goyal L, Shi L, Liu LY et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov, 2019; 9: 1064-1079.[DOI]
[16] Zhou Z, Chen X, Fu Y et al. Characterization of FGF401 as a reversible covalent inhibitor of fibroblast growth factor receptor 4. Chem Commun, 2019; 55: 5890-5893.[DOI]
[17] Chan SL, Schuler M, Kang YK et al. A first-in-human phase 1/2 study of FGF401 and combination of FGF401 with spartalizumab in patients with hepatocellular carcinoma or biomarker-selected solid tumors. J Exp Clin Canc Res, 2022; 41: 189.[DOI]
[18] Weiss A, Adler F, Buhles A et al. FGF401, A first-in-class highly selective and potent FGFR4 inhibitor for the treatment of FGF19-driven hepatocellular cancer. Mol Cancer Ther, 2019; 18: 2194-2206.[DOI]
[19] Wu X, Dai M, Cui R et al. Design, synthesis and biological evaluation of pyrazolo [3, 4-d] pyridazinone derivatives as covalent FGFR inhibitors. Acta Pharm Sin B, 2021; 11: 781-794.[DOI]
[20] Molecular Operating Environment (MOE) | MOEsaic | PSILO. Accessed December 2023. Available at:[Web]
[21] DUD-E: A Database of Useful (Docking) Decoys - Enhanced. Accessed December 2023. Available at:[Web]
[22] Mysinger MM, Carchia M, Irwin JJ et al. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem, 2012; 55: 6582-6594.[DOI]
[23] Specs - Compound Management Services and Research Compounds for the Life Science Industry. Accessed December 2023. Available at:[web]
[24] Sunseri J, Koes DR. Pharmit: interactive exploration of chemical space. Nucleic Acids Res, 2016; 44: W442-W448.[DOI]
[25] Hospital A, Goñi JR, Orozco M et al. Molecular dynamics simulations: advances and applications. Adv Appl Bioinf Chem, 2015; 8: 37-47.[DOI]
[26] Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. Bmc Biol, 2011; 9: 1-9.[DOI]
[27] Abraham MJ, Murtola T, Schulz R et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 2015; 1: 19-25.[DOI]
[28] Al-Karmalawy AA, Dahab MA, Metwaly AM et al. Molecular docking and dynamics simulation revealed the potential inhibitory activity of ACEIs against SARS-CoV-2 targeting the h ACE2 receptor. Front Chem, 2021; 9: 661230.[DOI]
[29] Zaki AA, Ashour A, Elhady SS et al. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies. J Tradit Compl Med, 2022; 12: 16-34.[DOI]
[30] Ross GA, Rustenburg AS, Grinaway PB et al. Biomolecular simulations under realistic macroscopic salt conditions. J Phys Chem B, 2018; 122: 5466-5486.[DOI]
[31] Hamed MIA, Darwish KM, Soltane R et al. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: Rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv, 2021; 11: 35536-35558.[DOI]
[32] Tuble SC, Anwar J, Gale JD. An approach to developing a force field for molecular simulation of martensitic phase transitions between phases with subtle differences in energy and structure. J Am Chem Soc, 2004; 126: 396-405.[DOI]
[33] Jones D, Allen JE, Yang Y et al. Accelerators for classical molecular dynamics simulations of biomolecules. J Chem Theory Comput, 2022; 18: 4047-4069.[DOI]
[34] Turner PX. Version 5.1.19. Cent Coast Land-Margin Res Oregon Grad Inst Sci Technol Beavert. Sci Rep-Uk, 2024; 14: 1152.[DOI]
[35] Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics, 1996; 14: 33-38.[DOI]
[36] Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics Poisson - Boltzmann surface area method. Mol Inform, 2012; 31: 114-122.[DOI]
[37] Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA et al. Gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J Chem Theory Comput, 2021; 17: 6281-6291.[DOI]
[38] Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem, 1999; 20: 217-230.[DOI]
[39] Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Dis, 2015; 10: 449-461.[DOI]
[40] Lagunin AA, Dubovskaja VI, Rudik AV et al. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. Plos One, 2018; 13: e0191838.[DOI]
[41] Opo FADM, Rahman MM, Ahammad F et al. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci Rep-Uk, 2021; 11: 4049.[DOI]
[42] Madden JC, Enoch SJ, Paini A et al. A review of in silico tools as alternatives to animal testing: principles, resources and applications. Altern Lab Anim, 2020; 48: 146-172.[DOI]
[43] Brogi S, Ramalho TC, Kuca K et al. In silico methods for drug design and discovery. Front Chem, 2020; 8: 612.[DOI]
[44] Mahfuz A, Khan MA, Biswas S et al. In search of novel inhibitors of anti-cancer drug target fibroblast growth factor receptors: Insights from virtual screening, molecular docking, and molecular dynamics. Arab J Chem, 2022; 15: 103882.[DOI]
[45] Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem, 2015; 58: 4066-4072.[DOI]
[46] Zhang Z, Wang Y, Chen X et al. Characterization of an aromatic trifluoromethyl ketone as a new warhead for covalently reversible kinase inhibitor design. Bioorg Med Chem, 2021; 50: 116457.[DOI]
[47] DeLano WL. The PyMOL molecular graphics system. Available at:[Web]
[48] Lipinski CA, Lombardo F, Dominy BW et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev, 1997; 23: 3-25.[DOI]
[49] Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem, 1999; 1: 55-68.[DOI]
[50] Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Reports, 2017; 7: 42717.[DOI]
Copyright © 2024 The Author(s). This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.


