Fluorescence Thermal Shift Assay based High Through-put Screening in Small Molecule Drug Discovery: A Brief Overview
Vinita Chauhan Kushwah1, Ritika Chauhan1, Ram Kumar Dhaked1*
1Biotechnology Division, Defence Research & Development Establishment, Gwalior, India
*Correspondence to: Ram Kumar Dhaked, PhD, Scientist, Biotechnology Division, Defence Research & Development Establishment Gwalior, 474002, India; Email: rkdhaked.drde@gov.in
Abstract
Background: The Early drug discovery process was majorly phenotypic, lacking target specificity, and mechanism of action causing short and/or long-term toxicological impacts on the patients leading to several drug withdrawals from the market. Biochemical procedures and methods used in drug discovery and development make it lengthy in terms of time requiring approx. 15 years with the investment of billions of dollars for a new drug molecule. Technological advancement leading to the identification of protein structures through crystallography paved a new path toward targeted drug discovery.
Objective: This paper provides a brief overview of the use of the fluorescence thermal shift assay (FTSA) as a high throughput (HTS) screening technique in small molecule drug discovery.
Methods: The article discusses the principles of thermal denaturation techniques and how they are applied in drug discovery. It highlights the advantages of FTSA over other biophysical methods, including its label-free, cost-effective, and high HTS nature. The review also presents a comparative analysis of FTSA with techniques such as isothermal titration calorimetry (ITC), mass spectrometry (MS), nuclear magnetic resonance (NMR), surface plasmon resonance (SPR), X-ray crystallography(X-RD), circular dichroism (CD), and differential scanning calorimetry (DSC).
Results: The FTSA technique is described as a powerful tool that addresses the limitations of larger protein requirements, specific assay development, and low HTS associated with other thermal denaturation methods. Unlike biochemical and cellular assays that measure secondary effects, FTSA directly estimates the binding of small molecules to target proteins.
Conclusion: The review concludes that FTSA is the only biophysical method that satisfies the three key prerequisites of being label-free, cost-effective, and suitable for high HTS screening in small molecule drug discovery.
Keywords: FTSA, CETSA, Tm, thermal stability, drug discovery, high through-put screening
1 INTRODUCTION
The Drug discovery process has been observed a long way since its inception in terms of procedural developments and methods involved. Early drug discovery approach was majorly phenotypic, lacking information regarding specific targets or mechanisms of action[1-3]. The limitation of such an approach posed unidentified short-term or long-term toxicological impacts on the patients. Selective binding with high affinity of the drug to the target is important to be both effective and safe. Insufficient target engagement and/or off-target interaction-induced toxicities are the main reasons for the high attrition rate in drug development. Side effects of drugs developed based on functional and phenotypic resulting from off-target pharmacology, identified with the advancements in biochemical pharmacology led to withdrawal from clinics. The technological development in identification of protein structures majorly through crystallography paved new avenues towards targeted drug discovery[4]. The structural biology combined with protein function discovery of various pathways emerged as a giant leap in drug development. This replaced the limited compounds evaluation in animal models with a targeted high throughput (HTS) screening of over a million compounds through biochemical assays. However, biochemical methods have their own limitations of reproducibility, requirement of expensive infrastructure and expertise and biochemicals. On the other hand, biophysical methods are instrument driven, thus reducing these limitations and providing quick reproducible results employing high through-put assay systems.
The role of biophysical methods in drug discovery is extensive, ranging from target identification to construct selection, ligand screening and optimization. These methods play a significant role in validation of hits obtained through biochemical and cell-based assays. Various biophysical techniques like fluorescence resonance energy transfer (FRET), thermal denaturation methods, mass spectrometry, nuclear magnetic resonance (NMR), surface plasmon resonance (SPR), X-ray crystallography(X-RD) and molecular imaging have become key components in the drug discovery process[5].
1.1 Thermal Denaturation Techniques in Drug Discovery
The thermal denaturation techniques are used for protein-ligand interaction studies wherealtered thermal stability of a protein is estimated in presence of substrates, cofactors, small molecules, metal ions and even other proteins[6-9]. Thermal stability or denaturation of proteins can be monitored through environmental sensitive fluorescence dye that enables the fast and inexpensive determination of melting temperature Tm using real-time PCR machines. The applications of thermal denaturation techniques in small molecule drug discovery are based on the fact that thermal stability of target proteins could be enhanced or reduced when a small molecule comes in close proximity that depends on small molecule binding affinity[10-12]. In the drug discovery process circular dichroism (CD), isothermal titration calorimetry (ITC) and differential scanning calorimetry (DSC) has been implemented to explore the thermal stability of target protein[13-15]. ITC is a label free technique that provides rich information on reactions and is considered as a “gold-standard” in pharmacology. However, instrumentation with automation set-up is expensive and at the best it is a low-to-medium through-put method. DSC is used for determination of energy required for phase transition of protein or melting. It is an excellent label-free technique but is time-consuming, requiring a dedicated expensive instrument that offers low through-put thus not suitable for screening applications. However, the high through-put automated application of thermal stability measurement of target proteins is achieved through fluorescence thermal shift assay (FTSA)[11,12]. FTSA is also known as ThermoFluor, differential scanning fluorimetry (DSF) or temperature dependent fluorescence[15]. This method is being adopted by drug discoverers that addresses the limitations of large amounts of protein requirement, specific assay development and low through-put as encountered in other thermal denaturation techniques[16-18]. Unlike biochemical and cellular assays where readout represents secondary effects like enzyme inhibition or second messenger production, direct binding is estimated in at scale using FTSA (Figure 1). A ready reference comparison of FTSA with different biophysical techniques has been drawn considering HTS, sensitivity and cost (Table 1).
|
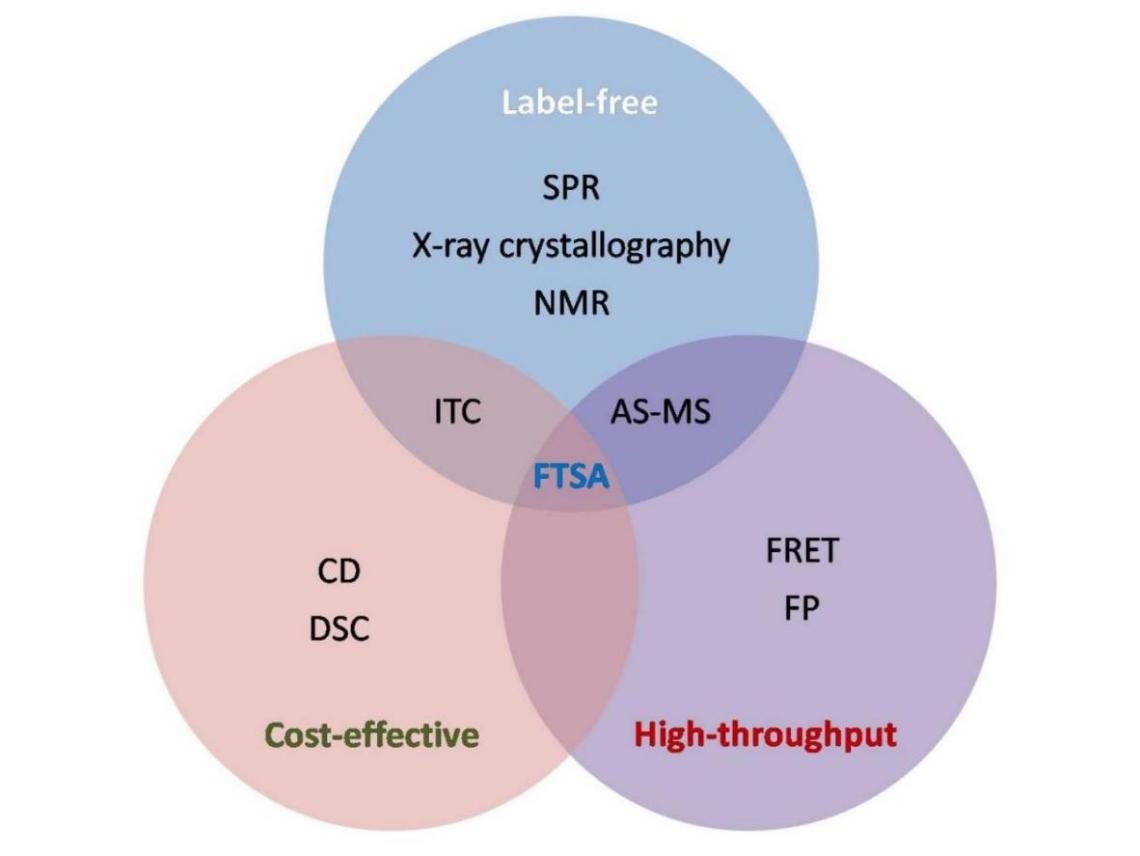
Figure 1. The Venn diagram of the interface of the three prerequisites used in drug discovery and development. The Venn diagram depicting the interface of the three prerequisites of label free, cost-effective and high through-put biophysical techniques used in drug discovery and development. The techniques considered are SPR, X-RD, NMR, CD, DSC, FRET, fluorescent protein, ITC, affinity selection mass spectrometry , and FTSA. The only method compliant with all three properties is FTSA.
Table 1. Comparison of FTSA with Other Biophysical Methods with Respect to Sensitivity, HTS and Cost
Technique |
Pros |
Cons |
ICT |
Label free |
Not suitable for HTS, large amount of sample needed |
MS |
Label free, sensitive |
High cost, require expertise |
NMR |
Label free |
Low sensitivity, high cost, require expertise, not suitable for HTS |
SPR |
Label free, highly sensitive |
Expensive, long calibration process |
X-RD |
Label free, |
Not used for HTS, low sensitivity |
DSC |
Cost effective, high sensitive |
Not suitable for HTS |
CD |
Cost effective |
Not suitable for HTS |
FRET |
Suitable for high through put, highly sensitive |
High cost |
FP |
Suitable for high through put |
High cost |
FTSA |
Label free, Cost effective, suitable for HTS, highly sensitive |
Required simple buffer not compatible with biological fluids |
1.2 Why FTSA?
Numerous approaches exist in drug discovery process for studying the interactions between target and ligands based on fluorescence or enzyme assays. However, labelled probes not only raises costs but limits conditions and the tested compounds might interfere with label, leading to false positive screening candidates. A major advantage of FTSA, utilize the thermodynamic coupling between ligand and bio-molecule, unfolding applying established thermodynamic relationships to describe the binding properties[11,12]. FTSA quantifies the shift in the melting temperature of the target in real-time upon binding of ligands and compounds that significantly shift in thermal melting of protein are identified as true binders. Assay has several advantages over other biophysical methods being cost effective and simplest to perform high through-put ligand screening (Figure 2). This assay does not require a huge capital instrument, expensive biochemicals and accomplished through much simpler procedure. Application of assay is not only limited to hit confirmation but also used as primary screening assay in fragment-based drug discovery (FBDD)[16,17]. The inhibitor screening through FTSA is characterized by change in midpoint for melting curves ∆Tm of ligand-protein complex relative to un-complexed protein due to energy linkage between ligand and protein. Ligand-protein binding is determined via shift of protein unfolding temperature; however, this assay is unable to provide direct information regarding location of the interaction and structure[19].
|
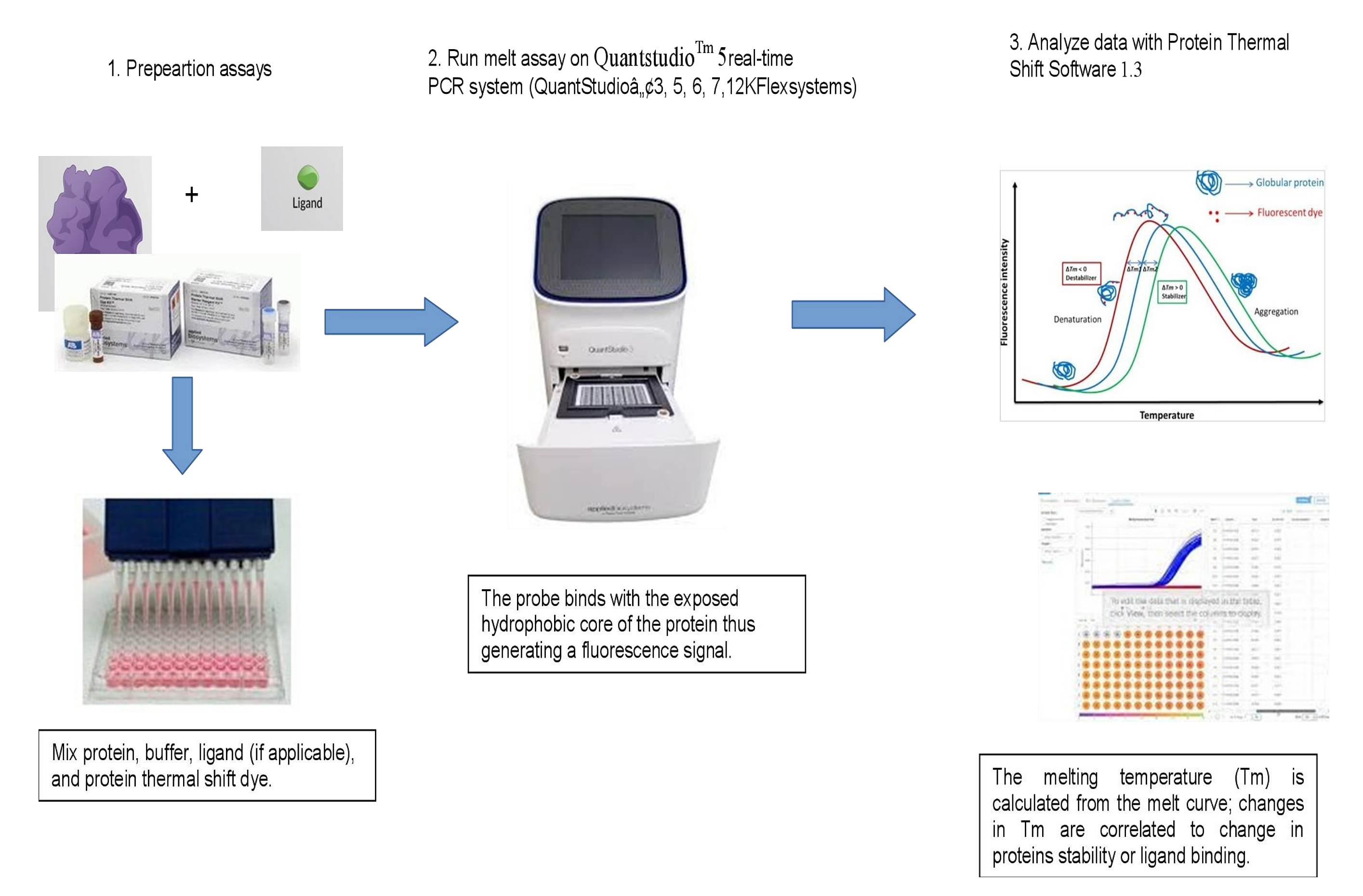
Figure 2. Schematic representation of FTSA workflow.
1.3 FTSA Principle
An environment sensitive fluorescent probe molecule, most commonly SYPRO Orange, is used for thermal shift quantification. When a protein in an aqueous medium is subjected to increasing temperatures there is break in non-covalent bonds that are responsible for protein folding. This results in hydrophobic amino acids of soluble proteins being disproportionately sequestered and exposed upon unfolding. The probe binds with the exposed hydrophobic core of the protein thus generating a fluorescence signal that increases with increasing non-polar environment. However, with further heating the protein is completely denatured and begins to aggregate and the fluorescence signal drops due to precipitation of protein-dye complex. The temperature at which the unfolding of half of the protein is attained is termed as ‘melting point temperature’ or Tm. In presence of a ligand the protein-ligand complex is formed which might increase or decrease the stability of protein and when this complex is subjected to heat a shift in Tm is observed[19,20] (Figure 3).
|
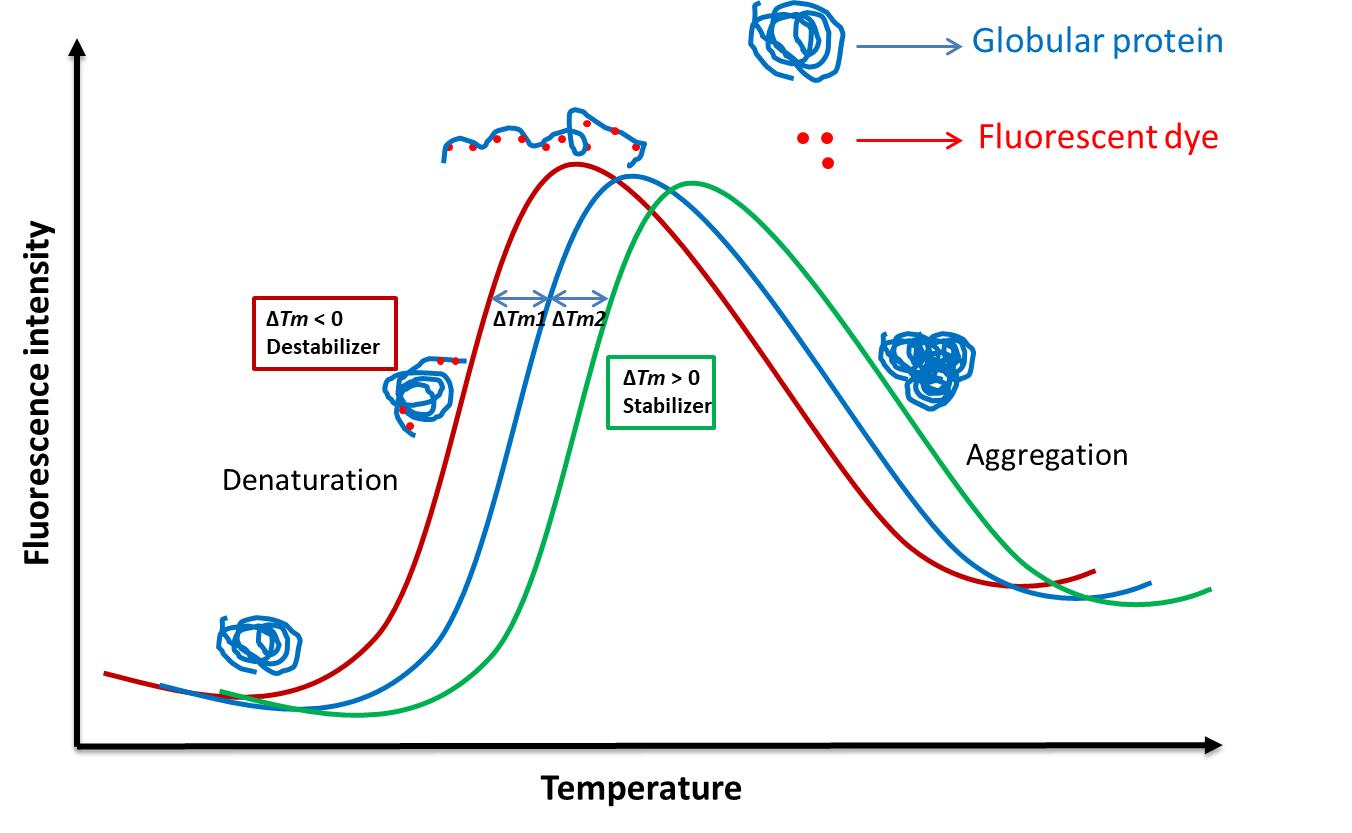
Figure 3. The melt curve of changes in Tm with temperature in the presence of the ligand.
In standard FTSA, in absence of a ligand the protein melting takes place with increase in temperature resulting in binding of the environment sensitive fluorescent dye with the hydrophobic patches of the unfolded protein. Heating leads to protein aggregation causing reduced fluorescence in the later stage (blue sigmoid curve). The temperature at which the protein is at equilibrium of folded and unfolded state is termed as Tm. In presence of a ligand the shift in Tm observed where the protein-ligand complex can be stabilized (green sigmoid curve, ∆Tm2) or destabilized (red sigmoid curve, ∆Tm1) which can be quantified through the melt curve.
The protein stability is reflected by ∆Gu i.e., difference in Gibbs free energy of the folded (Gf) and the unfolded states (Gu). The quantification of protein stability is performed through temperature dependent change in Gibbs free energy of unfolding (∆Gu). Consequently, when the protein stability is reduced with increase in temperature ∆Gu decreases, until it becomes zero at equilibrium position when equal concentrations of folded and unfolded protein are present[19-22]. Proteins are higher-order structures consisting of enthalpic (hydrogen bonds, hydrophobic interactions, salt bridges, etc. and entropic (rotation of side chains, loops flexibility, etc.) components. Denaturation of protein in presence of ligand is generally entropy driven phenomenon depending on their ability to reduce one or more entropic components. However, the binding enthalpy is comparatively smaller than melting enthalpy, hence may not be the main force behind thermal shift[21,22].
2 EXPERIMENTAL SETUP
The experimental setup for conducting FTSA as mentioned earlier is rather simple that comprises 5 basic components: (1) buffer, (2) protein, (3) fluorescent dye, (4) ligand, (5) real time PCR (RT-PCR) instrument[20].
2.1 Buffer Optimization
The buffer used for FTSA is relatively standard where any common near neutral buffer with minimal salts and additives could be used. The major concern is to minimize hindrance in the fluorescent signal and protein stabilization. Common buffers such as HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), phosphate and acetate are generally used for the process[20]. These buffers are characterized by high pH stability with increase in temperature thus generating clear melting transitions. Buffers of low or high PH with high salt content may provide erroneous results.
2.2 Protein Optimization
The amount of protein required for assay depends on its size, which may be optimized using 1.0 to 20µM concentrations and should not be unfolded or aggregated[20]. Usually, 1.0µM concentration can be used for most of the proteins and optimized up to 100nM for larger proteins. Purity of protein is paramount and the minimum purity required for FTSA is 75%, as impurities may give rise to broad or several fluorescence peaks. In general, the peak that shifts after ligand binding compared to peak observed without ligand is to be considered for estimation of Tm in such cases. The protein can be dialyzed or diluted in the reaction buffer to reduce the signal hindrance from any stabilizing additives. The Tm of protein too plays a significant role in obtaining the melting curve and should be between 30-90℃.
2.3 Fluorescent Dyes
Environment sensitive fluorescent dyes used in FTSA are characterized by low fluorescence in high dielectric constant medium and high fluorescence in low dielectric constant medium such as hydrophobic patches of unfolded proteins[23,24]. In early FTSA experimental setups the most commonly used dyes were 1-anilinonaphthalene-8-sulphonic acid (1,8-ANS), 2-anilinonaphthalene-6-sulphonic acid (2,6-ANS) and 2-(p-toluidinyl)-napthalene-6-sulphonic acid (2,6-TNS)[25-27]. These dyes however require UV excitation and quartz cuvettes, as the excitation and emission maxima are not easily reachable through commercial filters of RT-PCR machines. Presently, SYPRO Orange is the most frequently used dye having the excitation and emission maxima at 480 and 569nM, respectively. The range of concentrations from 0.5 to 10X is sufficient for optimization[20]. This dye typically undergoes large intensity change when it interacts with unfolded proteins and this wavelength range is easily obtained in RT-PCR instruments of different make[27]. Development of FTSA using these dyes for membrane proteins is rather challenging due to their high hydrophobicity with limited hydrophilic region and presence of detergents resulting in high fluorescence background. Thiol-specific dyes N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM), (CPM; λex398/λem482nM) or boron-dipyrromethene (BODIPY), (BODIPY FL-L-cysteine; λex505/λem512nM) are being used to study stability of membrane proteins in different solutions including ligand binding. BODIPY FL-l-cysteine has advantages over CPM dye due to its ability to minimize autofluorescence thus the false positive or negative results[24].
2.4 Ligand Optimization
The compound concentration is preferred within the kinetic solubility limits but excess with respect to protein concentration. The high ligand concentration is kept to ensure saturation of the entire binding sites. The standard ratio of ligand to protein concentration is 10:1 or higher. The ∆Tm is driven by free ligand rather than ligand bound to protein and hence higher concentration of ligand in the reaction mixture maintains the accuracy of ∆Tm measurements. Increasing solvents concentrations, such as DMSO dissociates disulfide bonds and destabilizes proteins resulting in hindrance in melting transition that requires either solvent exchange or dilution up to 2-5% v/v in the final reaction mixtures[10]. Also, DMSO at higher concentrations could lead to negative shifts in ∆Tm depending on the nature of the protein as reported by Andreotti et al[25].
2.5 Instrumentation
The most common instrument adapted for use in FTSA is real-time PCR machine consisting of the optical system compatible with fluorescent dyes. Most commonly used instruments are LightCycler480 from Roche[26], iCycler/iQ from BioRad[27], Mx3005P of Stratagene[28], Rotorgene from Corbet Research[29], FluoDia T70 from PTI and various real-time PCR machines of Applied Biosystems[30]. Most of these instruments are equipped with FTSA analysis software. The CFX96 a real-time PCR instrument from Bio-Rad Laboratories used curve-fitting software XLfit5 (ID Business Solutions Ltd.) for FTSA. Another instrument that can be used is a fluorescent plate reader with a heated stage.
2.6 Data Analysis
To avoid high fluorescence in no protein control and ligand only control due to contaminated protein, optimization wrt to dye and proteins is required. Each protein will require different optimal conditions. However, most proteins are successful with the first set of screens. Protein does not have enough hydrophobic residues to provide a strong fluorescent signal (i.e., small proteins), or has too many exposed hydrophobic residues, may result in high background fluorescence and experiment may fail if detergents are present in the buffer.
Generic tools for protocols and data analysis are available with the instrument for initial screening. In general, a positive ∆Tm is a result of stronger binding of ligand to the native folded protein than unfolded state (protein is stabilized) and the binding of ligand to the unfolded form is stronger than that of native state produces negative ∆Tm (protein is destabilized). Careful examination is required of raw data as the fit of this will provide Tm and the fit of Tm data will determine the Kd (dissociation constant or binding affinities). Despite the several applications of FTSA for studying protein-small molecule interactions, only limited studies have described determination of dissociation constants. Ligand-binding affinities Kd can be determined using assay, as an increase in Tm of the protein after ligand binding depends on the ligand concentration and binding affinity[31-35]. Intensity of fluorescence is plotted as a function of the temperature generating regular sigmoidal unfolding curve. In some cases, unusually shaped curves may result due to low temperature shifts, multiple transitions, high background fluorescence, and if ligand binding affects unfolding modes of the protein. Thermal unfolding may be modeled using Boltzmann or Derivatives models. Protein thermal shift software 1.3 calculates Tm-Boltzmann or Tm-Derivative to select the “positive hits”. The Tm is the inflection point of the sigmoid which is calculated using a Boltzmann equation which is used for identifying Tm and to report the presence/absence of binding (Equation (1)).
|
where, F is fluorescence intensity at temperature T. Fmax and Fmin are the fluorescence intensity values at upper and lower limits of the curve, respectively and a is the slope of the curve at Tm in a typical FTSA graph.
Most proteins provide consistent results using one or other method and are related to the shape of the unfolding curve. For determination of quantitative binding constants ‘thermodynamic models’ or ‘derivatives models’ have been used[33-35]. In the derivative method, it is important that multiple melting events are modeled. The derivative method takes the first derivative of the experimental data at each point and considers the melting temperature to be the point of highest first derivative. Matulis et al[31] included free ligand depletion consideration respective to the protein binding and equations for protein-ligand complex interaction have been modified. The ligand binding shifts the equilibrium towards native form. Equation (2):
|
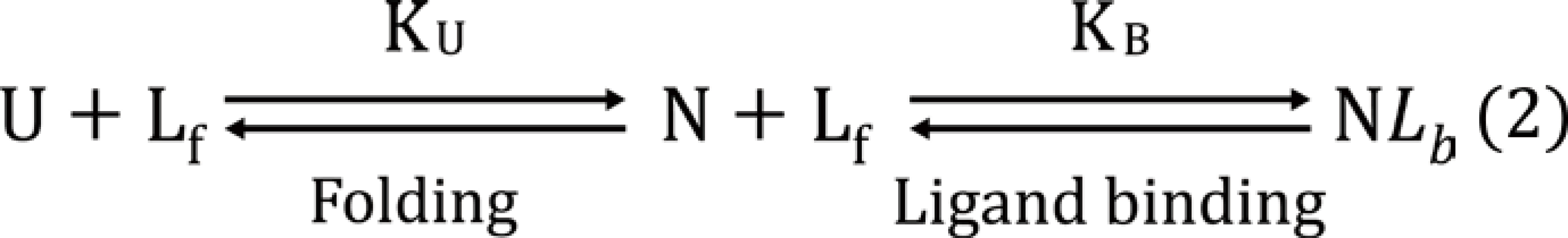
U = unfolded protein, N = native protein, Lf =free ligand, and NLb = native protein and ligand complex.
The Boltzmann equation was modified in the study as follows:
|
The equation for determination of Kb has been modified for ligand binding which was further employed for Kd determination.
|
Where,![]()
where yF,Tm and yU,Tm are fluorescence intensities in the presence of folded and unfolded protein at the melting temperature Tm, and mF and mU are the linear temperature dependences of fluorescence intensity plus folded and unfolded protein, respectively. The Gibbs free energy as a function of temperature (∆UG(T)) can be expressed in terms of the enthalpy (∆UHTr), entropy (∆USTr), and heat capacity (∆UCp) of protein unfolding at a reference temperature Tr, chosen to be Tm in the absence of ligand. The stoichiometry of the reaction can as well be determined using this equation with varying concentration of the ligand. These methods generally give a higher Tm of ~2-3℃. The ∆UHTr and ∆USTr are the Gibbs free energy and temperature dependent. Hence, it is expected that the Kd determined by FTSA is higher than that estimated at physiological temperature by other methods. Vivoli et al[36]. described detailed equations for determination of kinetic data of unfolding of the protein using many parameters.
The manual Tm analysis in case of high through-put screening is time-taking and excessive. There have been various free and commercial tools developed for data analysis including ThermoQ, LTSA, DMAN, Meltdown and MTSA[37-40]. TSA-CRAFT has been recently developed which provides a free, robust and cross-platform data analysis. This tool provides hassle free input file preparation, easy modifications and comprehensive Tm result display[41].
Principally in FTSA, Kd that is lower than 50% of the protein concentration is not possible to measure and at the upper end the sensitivity is determined by the solubility of the ligand. For the mathematically robust estimate of Kd, data have to be obtained with a 90% ligand-bound state of protein that requires ligand concentrations ~ 10X of Kd (considering no cooperativity). Hence, the limit of detection will be 1/10 of the solubility of the ligand[36].
3 FTSA IN HIGH THROUGH-PUT SMALL MOLECULE SCREENING
Therapeutic effect of small molecule-based drugs mostly mediated through their interactions with protein target and analysis of target engagement is a prerequisite objective in modern drug discovery methods. Development of screening method is a major challenge and resource intensive procedure for rapid identification of small molecule inhibitors of target proteins from compound libraries. Genomic projects lead in identification of potential new target proteins and is difficult to develop suitable functional assays to identify inhibitors for such uncharacterized targets. FTSA is one of the several approaches, that can be used for studying most types of protein receptor and small molecular weight ligand interactions. The assay has been reported to study interactions between different molecules ie, DNA, RNA, proteins, lipids, drugs, ions and other chemical entities. It is also reported in the identification of stabilizing buffers, osmolytes and additives that reduces unfolding or aggregation during purification and storage of proteins. FTSA can be used in sample preparation optimization for NMR and crystallization where high concentration of proteins is required for analysis[42]. FTSA being a time efficient, low cost, robust and versatile high through-put technique which is gaining popularity in the early stage of drug development. The major application of this assay is in high through-put screening of small molecule inhibitors against selected target proteins. This technique can also be used for validation of results obtained through other biochemical and biophysical assays. The considerations to obtain a visible melting curve, as discussed earlier, the temperature for the reaction is taken in the range of 25-99℃ with temperature ramp rate of 1℃ per minute. One can screen thousands of compounds for target specificity and affinity within an hour[21]. In general, addition of compounds shows dose-dependent increase in melting temperature of proteins and a temperature shift ∆Tm of over 2℃ is considered significant in screening of the compound libraries; however, less than 0.2℃ shift can be observed using advanced monitors[31].
FTSA has been used by Fedorov et al[33] to determine specificity of 156 validated kinase inhibitors against active and inactive human Ser/Thr kinases. The target specificity has also been evaluated by using off-targets such as post-translationally modified variants, mutants or truncated proteins for ligand interaction[43]. The major application of FTSA till date has been observed in the identification and validation of pharmacological chaperones[25]. The potential use of chaperones in identification and stabilization of protein mutants leading to proper cellular functioning has also been explored. The target protein used in the small molecule binding and activity assays should be as identical as possible. The influence of expression constructs and purification procedures has been explored by Rudolf et al[44]. They purified 15 kinases from E. coli and screened them against 244 known kinases. This screening displayed a significant increase in specificity of the compounds where 71% compounds showed IC50 value less than 0.5mM compared to previous study where 49% compounds were showing such inhibitory potential[45]. This signifies the importance of close identity of the kinases that were obtained through similar procedure. The method has been used for target identification and inhibition mechanisms of four reported antiviral drugs against Ebola virus infection and it was observed that the FTSA findings correlate well with protein-inhibitor interaction and antiviral activities[46]. FTSA has been employed for high through-put screening of small molecules against Shiga toxin and amide based small molecules were identified[30,40]. Recently, assay has been used for identifying small molecule inhibitors of botulinum neurotoxins and ribosome inactivating proteins[7,9].
This method can be also be used for discrimination of the molecules with undesired non-specific inhibitors such as fluorescence quenchers, promiscuous aggregators and redox cyclers known as PAINS (pan assay interfering compounds) during drug discovery and development[47]. FTSA may help in studying industrial enzymes that need sufficient stability towards the required reaction conditions such as low-high pH, high reactants concentration, and non-ambient temperatures. The method is also being used in monitoring the purification of native/recombinant proteins.
4 RECENT ADVANCEMENTS
Analyzing engagements between a drug and target protein in a physiologically relevant environment is a major challenge in the drug discovery process. FTSA is being performed using purified proteins and environment-sensitive dyes in a non-complex medium. Nanoscale DSF has been developed to estimate the intrinsic fluorescence of tryptophan (TRP)/tyrosine (TYR) residues which is strongly dependent on their local surroundings in 3D-structure of the proteins[48]. Unfolding of the proteins leads to change in intrinsic TRP/TYR fluorescence that translates into a shift in emission peak and intensity. The changes in fluorescence can reveal multiple unfolding transitions with high time resolution using NanoDSF monitors that are able to measure fluorescence intensity of tryptophan at two wavelengths i.e., 330 and 350nM. The cellular thermal shift assay (CETSA), is an advancement in thermal shift assay that enables to quantify the changes in the thermal stability of proteins in intact cultured cells and tissue explants for in situ assessment of drug target engagement. CETSA can be used for monitoring drug efficacy and safety as well as drug resistance at the target engagement, for instance during cancer therapy[49]. Dart et al.[50] reported NanoLuc luciferase thermal shift assay (NaLTSA) where protein targets appended with small luciferase (NanLuc) were used for small molecule screening and profiling. The assay can be performed in a complex environment with specificity rendered by tagging the folded state of the target protein to NanLuc without requiring specific antibodies or purified protein. These modified versions have several of advantages that include the ability to detect binding in cellular models without requiring purification or tagged target proteins, or probe compounds (Figure 4). All of these TSA variants may be applicable to determine thermal stability of both soluble and membrane proteins.
|

Figure 4. Modified versions of TSA. A. NanoDSF, B. CETSA, C. NaLTSA has a number of advantages including the ability to detect binding in cellular models without requiring purification or tagged target proteins, or probe compounds.
5 ADVANTAGES AND LIMITATIONS
FTSA displays significant advantages in small molecule drug discovery by being high HTS, inexpensive, reproducible and robust technique. This method utilizes a very small amount of receptor protein and has the minimal requirement of fluorescent dyes that can be obtained very easily. It is a biophysical assay that eliminates the requirement of designing and setting up of new assay protocols for novel target proteins or functional assay of reported protein drug targets. Another benefit of this technique is, it doesn’t require any prior knowledge of the protein function or protein-ligand interaction in order to determine the binding affinity.
Major limitations of FTSA that have been observed are the higher cases of false positive and negative results. The false positives are resulted from the non-specific interaction of the ligands or by the ligands interaction at multiple sites of the protein thus displaying higher ∆Tm. The cases of false negatives are observed when the protein is unstable or the compound has relative affinity with native and unbound states of the protein. A compound having a relatively low solubility or requirement of additional cofactor or metal ion cannot display the standard shift in Tm as well[51]. The requirement of fluorescent labels unlike ITC is another caveat where hydrophobic SYPRO Orange dye can compete with the hydrophobic ligands that may raise dissociation constant due to competition. Thermal stability studies of membrane proteins, encoded by 30% of the human genome and known drug targets using FTSA is still a challenge due to their high hydrophobicity and presence of detergents used for solubilization that increases considerable fluorescence background resulting in low signal-to-noise ratios.
6 FUTURE PERSPECTIVE
FTSA as a biophysical technique has an edge over its relatively expensive and low HTS counterparts (Table 1). Fragment-based ligand designing is hugely affected by advancements in biophysical techniques that can be used for primary and secondary screening of small molecules. With improved speed and sensitivity, the use of this method has gradually advanced in initial screening to identify new ligands in the drug discovery process by capitalizing on an increase in protein stability upon protein-ligand interaction. The fragment-based screening coupled with X-RD can provide detailed knowledge of fragments binding to the specific target that will facilitate the designing of molecules, by growing off or by combining different fragments, of required functionality. Its application in high HTS screening is quite distinguished despite minor drawbacks of false positives and negatives. The limitation can be overcome by performing other validation experiments in tandem/simultaneously with FTSA with a lesser number of selected molecules. CETSA has the potential to determine appropriate drug usage and dosage and to generate real-world data or Smart Data for the development of individualized drug treatment in the future. FTSA can also be used to analyze protein-protein interactions for studying disease -related misfolded proteins, and human protein interactome and will find applications for difficult multi-domain proteins. The assay can be integrated with computational inventions like artificial intelligence, machine learning, deep mining etc in predicting thermal shift upon interaction. Also, there is a scope to design highly sensitive new probes to make it experimentally robust to minimize pitfalls. In our opinion, FTSA has many advantages over the most biophysical techniques to be used in protein characterization and protein-ligand interaction in the near and far future.
Acknowledgements
Authors were thankful to Director DRDE, Gwalior. Ms. Ritika Chauhan (SRF) and Dr Vinita Chauhan (RA) were recipients of Fellowship from the Government of India. The manuscript was assigned laboratory accession No. DRDE/BT/18/2019. No external funding solicited for the work summarized in the manuscript.
Conflicts of Interest
The authors declared no conflicts of interest with respect to authorship for publication of this article.
Author Contribution
Dhaked RK was in charge of conceptualization, review and editing. Kushwah VC was in charge of writing original draft. Chauhan R was in charge of writing and editing. All authors read and approved the final version of manuscript.
Abbreviation List
1,8-ANS, 1-anilinonaphthalene-8-sulphonic acid
2,6-ANS, 2-anilinonaphthalene-6-sulphonic acid
2,6-TNS, 2-(p-toluidinyl)-napthalene-6-sulphonic acid
CETSA, Cellular thermal shift assay
CD, Circular dichroism
DSC, Differential scanning calorimetry
DSF, differential scanning fluorimetry
FBDD, fragment-based drug discovery
FRET, Fluorescence resonance energy transfer
FTSA, Fluorescence thermal shift assay
HTS, Throughput
ITC, Isothermal titration calorimetry
MS, Mass spectrometry
NaLTSA, NanoLuc luciferase thermal shift assay
NMR, Nuclear magnetic resonance
NanLuc, Small luciferase
SPR, Surface plasmon resonance
Tm, melting temperature
TRP, tryptophan
TYR, tyrosine
X-RD, X-ray crystallography
References
[1] Rees DC, Congreve M, Murray CW et al. Fragment-based Lead Discovery. Nat Rev Drug Disc, 2004; 3: 660-672.[DOI]
[2] Lipinski CA, Lombardo F, Dominy BW et al. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv Drug Del Rev, 2001; 46: 3-26.[DOI]
[3] Jencks WP. On the Attribution and Additively of Binding Energies. P Natl Acad Sci, 1981; 78: 4046-4050.[DOI]
[4] Erlanson DA, Fesik SW, Hubbard RE et al. Twenty Years on: The Impact of Fragments on Drug Discovery. Nat Rev Drug Disc, 2016; 15: 605-619.[DOI]
[5] Eggen M, Schindler J. Impact and Evolution of Biophysics in Medicinal Chemistry. Biophys Tech Drug Disc, 2017; 61: 1.[DOI]
[6] Lavis LD, Raines RT. Bright Building Blocks for Chemical Biology. ACSChemBiol, 2014; 9: 855-866.[DOI]
[7] Chauhan R, Chauhan V, Sonkar P et al. Targeted 8-hydroxyquinoline fragment based small molecule drug discovery against neglected botulinum neurotoxin type F. Bioorg Chem, 2019; 92: 103297.[DOI]
[8] Gonçalves MST. Fluorescent Labeling of Biomolecules with Organic Probes. Chem Rev, 2008; 109: 190-212.[DOI]
[9] Phatak P, Chauhan V, Dhaked RK et al. E-N-(2-acetyl-phenyl)-3-phenyl-acrylamide targets abrin and ricin toxicity: Hitting two toxins with one stone. Biomed Pharmacother, 2021; 143: 112-134.[DOI]
[10] Scott AD. Fluorescent Thermal Shift Assays for Identifying Small Molecule Ligands. Biophys Techn Drug Disc, 2017; 8: 320.[DOI]
[11] Vuorinen E, Valtonen S, Eskonen V et al. Sensitive label-free thermal stability assay for protein denaturation and protein–Ligand interaction studies. Analytical chemistry, 2020; 92: 3512-3516.[DOI]
[12] Cheng C, Liu M, Gao X et al. Identifying new ligands for JNK3 by fluorescence thermal shift assays and native mass spectrometry. Acs Omega, 2022; 7: 13925.[DOI]
[13] Kim HM, Cho BR. Small-molecule Two-photon Probes for Bioimaging Applications. Chem Rev, 2015; 115: 5014-5055.[DOI]
[14] Brandts JF, Hu CQ, Lin LN. A Simple Model for Proteins with Interacting Domains. Applications to scanning calorimetry data. Biochemistry, 1989; 28: 8588-8596.[DOI]
[15] Niesen FH, Berglund H, Vedadi M. The Use of Differential Scanning Fluorimetry to Detect Ligand Interactions that Promote Protein Stability. Nat Prot, 2007; 2: 2212-2221.[DOI]
[16] Terai T, Nagano T. Fluorescent Probes for Bio imaging Applications. Curr Opin Chem Biol, 2008; 12: 515-521.[DOI]
[17] Leitner PD, Vietor I, Huber LA et al. Fluorescent thermal shift-based method for detection of NF-κB binding to double-stranded DNA. Sci Rep, 2021; 11: 2331.[DOI]
[18] Bajar B, Wang E, Zhang S et al. A Guide to Fluorescent Protein FRET Pairs. Sensors, 2016; 16: 1488.[DOI]
[19] Redhead M, Satchell R, McCarthy C et al. Thermal Shift as an Entropy-driven Effect. Biochemistry, 2017; 56: 6187-6199.[DOI]
[20] Huynh K, Partch CL. Analysis of Protein Stability and Ligand Interactions by Thermal Shift Assay. CurrProtProtSci, 2016; 79: 28.9.1-28.9.14.[DOI]
[21] Pantoliano MW, Petrella EC, Kwasnoski JD et al. High-density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. J Biomol Screen, 2001; 6: 429-440.[DOI]
[22] Semisotnov GV, Rodionova NA, Razgulyaev OI et al. Study of the “Molten Globule” Intermediate State in Protein Folding by a Hydrophobic Fluorescent Probe. Biopol Orig Res Biomol, 1991; 31: 119-128.[DOI]
[23] Kean J, Cleverley, RM, O'Ryan L et al. Characterization of a CorA Mg2+ Transport Channel from Methanococcus jannaschii using a Thermofluor-based Stability Assay. Mol Memb Biol, 2008; 25: 653-661.[DOI]
[24] Hofmann L, Gulati S, Sears A et al. An Effective Thiol-reactive Probe for Differential Scanning Fluorimetry with a Standard Real-time Polymerase Chain Reaction Device. Anal Biochem, 2016; 499: 63-65.[DOI]
[25] Andreotti G, Monticellia M, Cubellis MV. Looking for protein stabilizing drugs with thermal shift assay. Drug Test Anal, 2015; 7: 831–834.[DOI]
[26] Breuer S, Chang MW, Yuan J et al. Identification of HIV-1 Inhibitors Targeting the Nucleocapsid Protein. J Med Chem, 2012; 55: 4968-4977.[DOI]
[27] Lo MC, Aulabaugh A, Jin G et al. Evaluation of Fluorescence-based Thermal Shift Assays for Hit Identification in Drug Discovery. Anal Biochem, 2004; 332: 153-159.[DOI]
[28] Egger S, Chaikuad A, Kavanagh KL et al. Structure and Mechanism of Human UDP - Glucose 6-Dehydrogenase. J Biol Chem, 2011; 286: 23877-23887.[DOI]
[29] Herzog G, Joerger AC, Shmueli MD et al. Evaluating Drosophila p53 as a Model System for Studying Cancer Mutations. J Biol Chem, 2012; 287: 44330-44337.[DOI]
[30] Chauhan V, Chaudhary D, Pathak U et al. In Silico Discovery and Validation of Amide Based Small Molecule Targeting the Enzymatic Site of Shiga Toxin. J Med Chem, 2016; 59: 10763-10773.[DOI]
[31] Matulis D, Kranz JK, Salemme FR et al. Thermodynamic Stability of Carbonic Anhydrase: Measurements of Binding Affinity and Stoichiometry using Thermo Fluor. Biochemistry, 2005; 44: 5258-5266.[DOI]
[32] Zubriene A, Matuliene J, Baranauskiene L et al. Measurement of Nanomolar Dissociation Constants by Titration Calorimetry and Thermal Shift Assay-Radicicol Binding to Hsp90 and Ethoxzolamide Binding to CAII. Inz J Mol Sci, 2009; 10: 2662-2680.[DOI]
[33] Fedorov O, Marsden B, Pogacic V et al. A Systematic Interaction Map of Validated Kinase Inhibitors with Ser/Thr Kinases. Proc Nat Acad Sci, 2007; 104: 20523-20528.[DOI]
[34] Cimmperman P, Baranauskiene L, Jachimoviciute S et al. A quantitative model of the thermal stabilization and destabilization of proteins by ligands. Biophys J, 2008; 95: 3222-3231.[DOI]
[35] Nan Bai N, Roder H, Dickson A et al. Isothermal Analysis of Thermo Fluor Data can readily provide Quantitative Binding Affinities. Scient Rep, 2019; 9: 2650.[DOI]
[36] Vivoli M, Novak HR, Littlechild JA et al. Determination of Protein-ligand Interactions Using Differential Scanning Fluorimetry. J Vis Exp, 2014; 91: 51809.[DOI]
[37] Phillips K, dela Pena AH. The Combined Use of the Thermo fluor Assay and Thermo Q Analytical Software for the Determination of Protein Stability and Buffer Optimization as an aid in Protein Crystallization. Curr Protoc Mol Biol, 2011; 10: 10-28.[DOI]
[38] Rosa N, Ristic M, Seabrook S A et al. Meltdown: A Tool to Help in the Interpretation of Thermal Melt Curves Acquired by Differential Scanning Fluorimetry. J Biomol Screen, 2015; 20: 898–905.[DOI]
[39] Schulz MN, Landstrom J, Hubbard RE. MTSA—A Matlab Program to Fit Thermal Shift Data. Anal Biochem, 2013; 433: 43–47.[DOI]
[40] Sviben D, Bertoša B, Hloušek-Kasun A et al. Investigation of the thermal shift assay and its power to predict protein and virus stabilizing conditions. J Pharmaceut Biomed, 2018; 161: 73-82.[DOI]
[41] Lee PH, Huang XX, Teh BT et al. TSA-CRAFT: A Free Software for Automatic and Robust Thermal Shift Assay Data Analysis. SLAS Discov, 2019; 24: 606-612.[DOI]
[42] Bruce D, Cardew E, Freitag-Pohl S et al. How to Stabilize Protein: Stability Screens for Thermal Shift Assays and Nano Differential Scanning Fluorimetry in theVirus-xProject. JVis Exp, 2019; 144.[DOI]
[43] Koszelak-Rosenblum M, Krol AC, Simmons DM et al. His-311and Arg-559 are Key Residues Involved in Fatty Acid Oxygenation in Pathogen-Inducible Oxygenase. J Biol Chem, 2008; 283: 24962-24971.[DOI]
[44] Rudolf AF, Skovgaard T, Knapp S et al. A Comparison of Protein Kinases Inhibitor 469 Screening Methods using both Enzymatic Activity and Binding Affinity Determination. PLoS One, 2014; 9: e98800.[DOI]
[45] Anastassiadis T, Deacon SW, Devarajan K et al. Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nat Biotech, 2011; 29: 1039-1045.[DOI]
[46] Ren J, Zhao Y, Fry EL et al. Target Identification and Mode of Action of Four Chemically Divergent Drugs Against Ebola virus Infection. J Med Chem, 2018; 61: 724-733.[DOI]
[47] Redhead M, Satchell R, Morkunaite V et al. A Combinatorial Biophysical Approach; FTSA and SPR for Identifying Small Molecule Ligands and PAINS. Anal Biochem, 2015; 479: 463.[DOI]
[48] Magnusson AO, Szekrenyi A, Joosten HJ et al. Nano DSF as Screening Tool for Enzyme Libraries and Biotechnology Development. FEBS J, 2018; 286: 184-204.[DOI]
[49] Molina DM, Nordlund P. The Cellular Thermal Shift Assay: A Novel Biophysical Assay for in situ Drug Target Engagement and Mechanistic Biomarker Studies. Ann Rev Pharm Toxicol, 2016; 56: 141-161.[DOI]
[50] Dart ML, Machleidt T, Jost E et al. Homogeneous Assay for Target Engagement Utilizing Bioluminescent Thermal Shift. ACS Med Chem Lett, 2018; 9: 546-551.[DOI]
[51] Hubbard RE, Murray JB. Experiences in Fragment-based Lead Discovery. Meth Enzymol, 2011; 493: 509-531.[DOI]
Copyright© 2024 The Authors. This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.


