Conotoxins: A Pharmacological Arsenal Yet to be Fully Explored
Aidan J. Ward1, K. H. Mok1*
1Trinity Biomedical Sciences Institute (TBSI), School of Biochemistry & Immunology, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
*Correspondence to: K. H. Mok, Trinity Biomedical Sciences Institute (TBSI), School of Biochemistry & Immunology, Trinity College Dublin, The University of Dublin, Dublin 2, D08 T9J0, Ireland; E-mail: mok1@tcd.ie
DOI: 10.53964/id.2025008
Abstract
Conotoxins are bioactive peptides derived from the venom of cone snails. Since its first biochemical characterization approximately 50 years ago, they have displayed a wide variety of neuronal and neuromuscular target specificities. Despite extensive research, conotoxins continue to remain a potential goldmine for pharmacology research and drug discovery. Their precise modulation of ion channels, receptors, and proteins positions them as promising candidates for treating chronic pain, diabetes, cardiovascular diseases, and neurological disorders. Of particular relevance is their role in addressing the current American opioid crisis, with conotoxins like ω-MVIIA (marketed as Prialt®) offering effective, non-addictive pain relief, showcasing their potential as alternatives to traditional opioids. Conotoxins also hold immense potential as blueprints for novel pharmaceuticals, but challenges remain in translating these peptides into clinical use. We provide a comprehensive literature review, focusing on how these peptides selectively interact with physiological targets located primarily in the nervous system. We chart the evolution of conotoxin research, highlighting significant progress and discussing key conotoxins like ω-MVIIA, Contulakin-G, α-RgIA4, mini-Ins, κO-PVIIA and αO-GeXIVA. Disulfide bridge pairings and sequences are included from ConoServer, the database of conotoxins, providing a comprehensive overview of the current research. After discussing the future application of AI in conotoxin research, advancements in computational modelling, and the importance of conservation efforts, we emphasize the need for continuing research to harness nature’s pharmacological arsenal fully and to set the stage for coming breakthroughs.
Keywords: conotoxins, drug discovery, nicotinic acetylcholine receptors, ion channels, pharmacology
1 INTRODUCTION
In the realm of natural venoms, few have captivated scientific interest as the venom of cone snails. These marine predatory molluscs from the genus Conus (recently redefined as Conidae) have evolved over millions of years to become one of the most sophisticated and deadly hunters in the oceanic realm. Their venom, a complex cocktail of toxic bioactive peptides, is a weapon for prey capture and a protective agent. Among these, conotoxins stand out as a group with exceptional diversity and pharmacological potential.
Cone snails, inhabiting tropical and subtropical oceans, boast around 1,000 species[1]. Despite their slow movement, they have developed the use of radular teeth as venom-injecting projectiles (Figure 1). These hollow, harpoon-like teeth, laden with venomous conotoxins, are ejected via an extendable proboscis, delivering this potent cocktail to their target[2]. Each cone snail species possesses a distinct conotoxin profile with a specific amino acid composition and pharmacological effects. Cone snail species are also separated by their diet: piscivorous (fish-hunting), molluscivorous (mollusc-hunting), and vermivorous (worm-hunting).
It is estimated that each cone snail species can synthesise between 1,000 and 1,900 different toxins, with minimal overlap of 5% between species[3]. Each conotoxin is tailored to a specific target. Most conotoxins act on the central and peripheral nervous system, often homing in on a narrow range of channel or receptor subtypes or even a specific subtype isoform in their prey. This remarkable precision of conotoxins in target specificity carries profound implications for drug discovery and pharmacology research. This is because many conotoxins are designed as a defence mechanism against humans and because of the physiological resemblance on the receptor level between their vertebrate fish prey and humans[3,4]. Yet, despite an estimated 1 million distinct conotoxins thought to exist, only about 80,000 have been identified, and even fewer have been pharmacologically characterised thoroughly[1,3].
|

Figure 1. C. purpurascens Captures a Clownfish. This chronological sequence begins with the clownfish stung by a barbed, hollow, harpoon-like radula tooth emanating from the tip of the snail’s proboscis (upper left). This rapid action injects venom through the tooth, anchoring the fish to the snail’s proboscis (upper right). Within 2 seconds, the venom takes effect, immobilising the fish (lower left). The immobilised fish is drawn into the snail’s rostrum and completely engulfed (lower right) Reproduced from Ref.[5] with permission from Springer Nature.
The discovery and isolation of conotoxins emerged in the latter half of the 20th century[6-12]. This was marked by a critical breakthrough with the isolation and characterization of the α-conotoxin GI from the venom of C. geographus, unveiling its structure and mode of action[13,14]. This pivotal discovery opened a new frontier in neuropharmacology as researchers began to decipher how this toxin could be harnessed to probe and manipulate the functioning of muscular contraction. These revelations sparked an enduring interest in the field, which has steadily grown and evolved over the past four decades. Initial research focused on pain management, with a landmark achievement in 2004 being the U.S. Food and Drug Administration (FDA) approval of Prialt® (synthetic ω-MVIIA), derived from the venom of the fish-hunting C. magus species, marking a transformative step in the clinical application of conotoxins[15,16].
Prialt® is distinguished by its ability to deliver precise, targeted analgesia with minimal off-target effects with proper dosing, setting it apart from many synthetic pharmacologic agents[15]. Additionally, it has no addictive properties[17]. This precision and efficacy have become particularly critical in addressing the opioid crisis, which stands as the most severe drug epidemic in the history of the United States[18,19]. The urgent demand for effective, non-addictive alternatives to opioids, which Prialt® and other conotoxins can provide, highlights the importance of continued research into conotoxins[16]. With this potential to provide potent analgesia without the risks of dependency or abuse, conotoxins hold significant promise for mitigating this widespread public health crisis, among others, which will be further clarified in detail in this review[1,3].
Structurally, conotoxins are small gene-derived bioactive peptides of usually 8 to 30 amino acid residues exhibiting vast diversity and complex structures. Post-translation modifications (PTMs) like C-terminal amidation, hydroxylation, and glycosylation enhance conotoxin chemical diversity. Conotoxins are notably characterised by their multiple disulfide bonds, which result in compact cysteine loop structures and impart exceptional structural stability. These structures frequently adopt α-helices and β-sheets, secondary structural elements typically observed in larger proteins. The unique structure of conotoxins is pivotal to their ability to bind with high affinity and specificity to a broad range of channels and receptors. Unlike many synthetic drugs, conotoxins have precise targeted action and minimal off-target effects[20].
In summary, conotoxins hold immense pharmacological potential, offering highly potent analgesics that provide pain relief surpassing morphine and addressing critical health challenges, such as the opioid crisis, while paving the way for innovative therapeutic approaches to various conditions. However, transforming conotoxins from venom-derived peptides to a viable drug has undergone significant challenges, including issues of stability, bioavailability, toxicity, and collapse of patent holders, which demand advanced research, technology, and patience to overcome[5]. Despite these obstacles, conotoxins represent a promising frontier in drug development, showcasing how biodiversity and evolutionary ingenuity can drive transformative advancements in medicine.
2 CONOTOXIN STRUCTURE, CLASSIFICATION AND FRAMEWORKS
2.1 Conotoxin Structure
Conotoxins are synthesised as messenger RNA (mRNA) precursor peptides, comprising distinct structural domains: a conserved signal peptide region, a propeptide region, and a cysteine-rich mature peptide region[21]. The signal peptide region, constant across a conotoxin gene superfamily, facilitates the secretion of the precursor peptide. The propeptide region aids in the intracellular processing of the toxin and precedes the mature peptide region, which ultimately embodies the bioactive component of the cone snail’s venom[22].
The discovery and identification of recently discovered conotoxins have been propelled by next-generation sequencing (NGS) technologies, which analyse the venom duct transcriptome, revealing the genomic bedrock of this pharmacological arsenal[23,24]. Conotoxins are classified into gene superfamilies based on the homology observed in their consensus signal sequences[25]. Despite this homology, the mature peptide regions within a superfamily exhibit substantial structural and functional variability. This diversity allows for a wide selection of target specificities and modes of action.
2.2 Conotoxin Classification
Conotoxin classification adheres to the nomenclature established by International Union of Basic and Clinical Pharmacology) (Figure 2), which organises these toxins based on their gene superfamilies and respective conotoxin frameworks[26,27]. There are 28 recognised gene superfamilies, each further categorised by the framework they employ[26,28]. Naming a toxin begins when the pharmacological properties become understood; once that happens, a Greek letter designation is incorporated into its nomenclature[26]. For instance, the designation “α-GIA” signifies an “α” conotoxin which targets the nicotinic acetylcholine receptors (nAChR), with “G” denoting its origin from C. geographus and “I” indicating the specific framework category[3,25,27,28]. Variants within these categories are often marked by letters such as “A”. When the pharmacological target of a conotoxin is unidentified, the Greek letter is excluded, and lowercase letters or a numeral (if singular) for variants, as shown by the terminology “ba32.1”. As illustrated in Table 1, the diversity of the conotoxin gene superfamilies within specific pharmacological classes, exemplified by α-conotoxins, necessitated a modified nomenclature. For instance, if an α-conotoxin falls outside the standard α-conotoxin superfamily (“A”) and belongs to a different one (let’s say “O1”, which GeXIVA is part of), it would be designated with an additional letter “O” following the “α”, such as “αO-GeXIVA”.
|
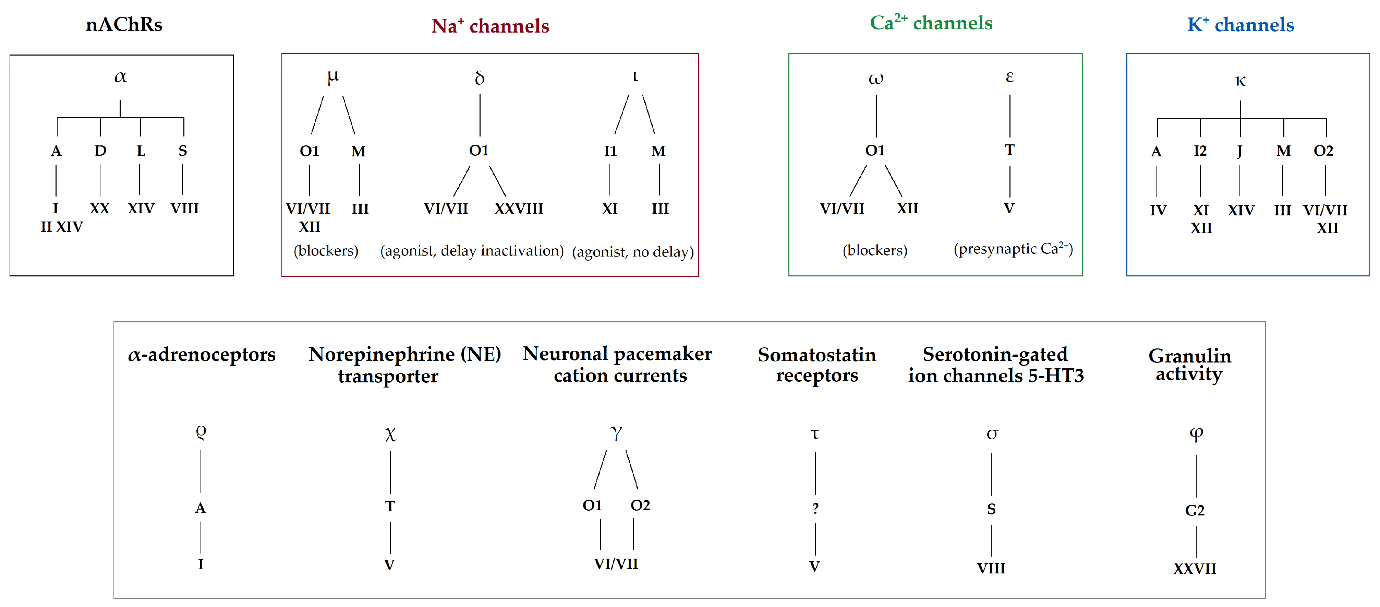
Figure 2. The conotoxin families are complex, and their classification has evolved. The focus has been on separating these peptides into Gene Superfamilies, which are classified based on their disulfide framework patterns. This classification is strongly correlated with pharmacological targeting due to the crucial role these frameworks play in stabilising the 3-D structure of the peptides. Reproduced from Ref.[29] with permission from MDPI.
Table 1. Representative Amino Acid Sequences for Some of the Known Conotoxin Pharmacological Families[27]
Superfamily (ies) |
Pharmacological family |
Reference Conotoxin |
Sequence |
Cysteine framework (s) |
Mode of Action |
A (B3, C, D, M, J, L, S, O1) |
α (alpha) |
GI |
|
A: I, II, IV, XIV M: III D: XX J: XIV S: VIII |
nAChR antagonist[30,31] |
A |
ρ (rho) |
TIA |
|
I |
α1A-adrenoreceptor inhibitor[32] |
T |
χ (chi) |
MrIA |
|
X |
NET inhibitor[33] |
τ (tau) |
CnVA |
|
V |
SSTR antagonist [34] |
|
O1 |
δ (delta) |
PVIA |
|
VI/VII |
Delay Nav inactivation[35] |
ω (omega) |
GVIA |
|
|
Cav blocker[36] |
|
O1 (M, A, I2, J) |
κ (kappa) |
PVIIA |
|
O1: VI/VII M: III A: IV I: XI/XII J: XIV |
Kv blocker[37] |
O2 |
γ (gamma) |
PnVIIA |
|
VI/VII |
Pacemaker channel activator[38] |
I1 (M) |
ι (iota) |
RXIA |
|
I1: XI M: III |
Nav activator[39] |
M (T) |
µ (mu) |
GIIIA |
|
M: III, IV T: V |
Nav blocker[40] |
G2 |
Φ (phi) |
MiXXVIIA |
|
XXVII |
Promotes cell proliferation[41] |
S |
σ (sigma) |
GVIIIA |
|
VIII |
HTR3A inhibitor [42] |
Insulin |
Con-insulins |
Cons-Ins G1 |
|
NA |
Insulin receptor agonists[43] |
NA |
Conopressins |
Conopressin-T |
|
Conopressin |
Vasopressin antagonists & agonists[44] |
Consomatins |
Consomatin pG1 |
|
Consomatin |
Selectively activates SSTRs[45] |
|
C |
Contulakins |
Contulakin-G |
ZSEEGGSNAτKKPYIL |
NA |
Cav inhibitor[46] |
Notes: PTMs are shown in blue bold: Z, pyroglutamate; O, 4-hydroxyproline; B, bromotryptophan; γ, γ-carboxyglutamate; τ, glycosylated threonine; *, C-terminal carboxamide; w, D-tryptophan; f, D-phenylalanine; Primary superfamily highlighted in bold where there are multiple; Cys residues involved in disulfide bonding are shown in red bold; Cys linkages in black.
2.3 Conotoxin Cysteine Frameworks
A defining feature of conotoxins is their cysteine frameworks, characterized by the specific number and arrangement of cysteine residues. Cysteine residues form disulfide bridges, which are crucial for the structural integrity of the conotoxins. The formation of disulfide bonds imparts a stable 3-D structure to the conotoxins, often leading to the formation of characteristic motifs. This structural stability is vital for their interaction with specific targets. In investigating novel sequences in cone snail venoms, a primary focus is deciphering the functions of these sequences by analysing their cysteine frameworks.
The ConoServer database has catalogued a comprehensive taxonomy of conotoxin cysteine framework families, with 33 currently separated by their unique cysteine patterns, loop sizes, and disulfide bond configurations[27]. Members of a conotoxin family share a high percentage of sequence similarity in their signal peptide sequence but less so in their propeptide sequence. As mentioned, they can be highly varied in their mature peptide sequence[47]. Framework I (α-conotoxins), Framework II (μ, ψ-conotoxins), and Framework VI/VII (ω, κ, μO, δ-conotoxins) have the largest number of members[48]. (See Figure 3 for a concise summary of known examples.)
|
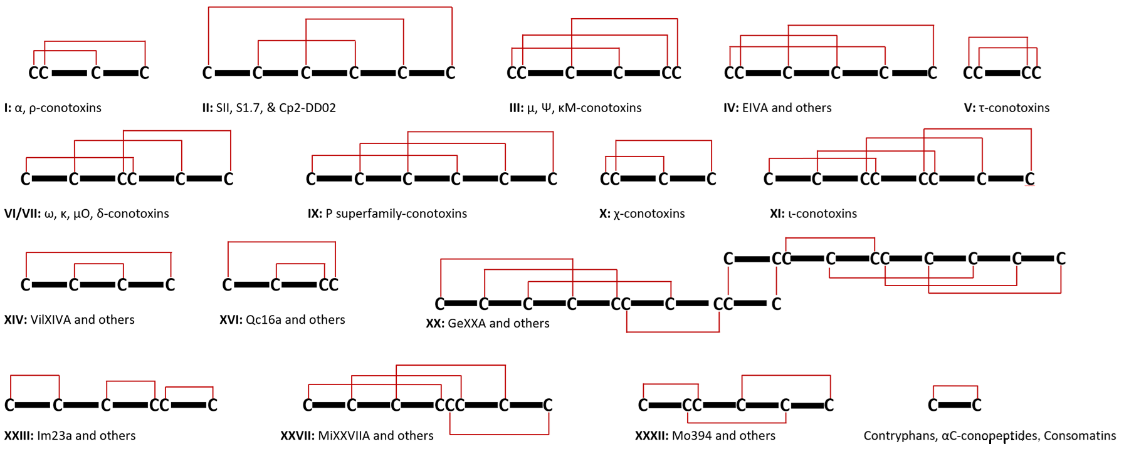
Figure 3. Illustrations of Cysteine Arrangements and Disulfide Bridges in Conotoxins Are Provided as Examples. It is crucial to recognise that the same cysteine patterns can result in various disulfide structures, complicating the prediction of disulfide connections in specific scenarios. Beyond these examples, there are numerous other known cysteine patterns whose disulfide linkages have not yet been established.
2.3.1 Framework I, CC-C-C
Framework I is the common cysteine framework for α-conotoxins observed in most Conus venoms. Key representatives of Framework I include α-Conotoxin ImI from C. imperialis and α-Conotoxin MII from C. magus, which has been used to differentiate and show receptor subunits in many physiological and pharmacological studies and recently are being applied towards targeting many conditions either as therapies or as pharmaceutical blueprints (Figure 4).
|
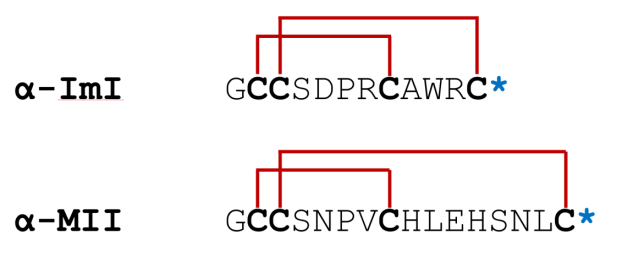
Figure 4. Arrangement of chosen Framework I conotoxins for comparison. Cysteine residues are marked in black, disulfide linkages are highlighted in red, and a star symbol denotes the C-terminus. Sequence post-translational modifications are indicated in blue bold.
2.3.1.1 α-ImI
α-ImI potently inhibits neuronal α7 nAChRs (IC50 is 497nM)[49,50]. Intracerebral administration of α-ImI in mice and rats induces complex seizures mirroring the effects of α-bungarotoxin, another α7 nAChR inhibitor. Discovered in 1994 and identified using high-performance liquid chromatography (HPLC), α-ImI was initially shown to block postsynaptic nAChRs in frog muscle fibres and was first shown to induce seizures in mice at higher doses[49].
Despite selectively inhibiting α7 nAChRs, α-ImI lacks analgesic activity[51]. This led to exploring its potential in cancer therapy as a blueprint or part of a targeted drug delivery system. α-ImI-modified micelles (ImI-PMs) were developed for targeted drug delivery to α7 nAChR-overexpressing tumors. These micelles, designed to carry paclitaxel, demonstrated enhanced targeting of breast cancer cells (MCF-7), leading to increased cellular uptake, cytotoxicity, and apoptosis[52]. In vivo studies showed improved tumour accumulation, reduced systemic toxicity, and enhanced anti-tumour efficacy compared to conventional paclitaxel formulations[52]. This established α-ImI as a promising ligand for developing targeted drug delivery systems for various α7 nAChR-expressing cancers. A further study demonstrated the efficacy of α-ImI-modified micelles in delivering docetaxel to non-small cell lung cancer (NSCLC), inhibiting tumour growth in α7 nAChR-overexpressing cells[53].
This was then followed up by a recombinant fusion protein study, ImI-AFP3, which was developed by combining α-ImI with alpha-fetoprotein domain 3 (AFP3), which inhibits the effects of high alpha-fetoprotein expression observed in hepatoid adenocarcinoma of the lung (HAL)[54,55]. ImI-AFP3 effectively inhibited the growth and migration of lung cancer cells cells. Furthermore, it significantly enhanced the anti-cancer activity of Gefitinib, a tyrosine kinase inhibitor, by overcoming resistance mechanisms. This synergistic effect increased apoptosis in lung cancer cells, particularly those with high AFP expression[54]. These findings demonstrate the potential of combining traditional drugs with bioengineered conotoxins like ImI to enhance their therapeutic efficacy.
2.3.1.2 α-MII
α-Conotoxin MII, known for potently inhibiting many nAChRs but not causing paralysis in fish or other vertebrates[56]. α-MII complements the role of α-ImI in the peripheral nervous system. While α-ImI inhibits rapidly decaying synaptic currents, α-MII targets the slower decaying currents[57]. α-MII has been widely employed in neurophysiology research as a selective inhibitor and molecular probe[58-62].
The α6* nAChR-selective antagonist analog MII[H9A; L15A] (IC50 2.4 nM for rat α6* nAChRs) has been employed to investigate the role of α6-containing nAChRs (α6* nAChRs) in nicotine addiction and has shown them as key players in mediating nicotine’s rewarding and addictive properties[62-64]. α6* nAChRs are predominantly expressed in brain regions for reward processing locations, such as the ventral tegmental area (VTA), where they play a pivotal role in nicotine’s rewarding effects and withdrawal symptoms, making them a potential therapeutic target for treating nicotine dependence[65]. Pretreatment with MII[H9A; L15A] effectively blocked nicotine-induced conditioned place preference (CPP), demonstrating the critical role of α6* nAChRs in mediating nicotine’s reinforcing effects[63]. Conversely, MII[H9A; L15A] did not significantly impact acute nicotine-induced analgesia, hypothermia, or locomotor activity, suggesting a limited involvement of α6* nAChRs[63].
In withdrawal assessments, anxiety-like behaviours were observed in the elevated plus maze and conditioned place aversion (CPA). However, MII[H9A; L15A] did not significantly influence physical withdrawal signs such as somatic symptoms or hyperalgesia, indicating a selective role for α6* nAChRs in mediating affective, but not physical, withdrawal manifestations[63]. These findings suggested that α6* nAChRs contribute to nicotine dependence primarily through the modulation of dopamine in the VTA. The locus coeruleus (LC), a brain region involved in stress responses, may also contribute to withdrawal-related effects mediated by these receptors[63,66]. Notably, blocking CPA required higher doses of MII[H9A; L15A] compared to blocking CPP, suggesting distinct underlying mechanisms for nicotine reward and withdrawal processes[63].
Furthermore, MII[H9A; L15A] has been used in highlighting the key roles of α6β2* and α4β2 nAChRs in nicotine and cocaine reward systems, with α6β2* nAChRs playing a central role in both[67]. This was done using a conditioned place preference (CPP) paradigm with pharmacological and genetic approaches[67]. Nicotine-induced CPP was absent in α6 knockout (KO) mice at moderate doses (0.5 mg/kg), but higher doses (1 mg/kg) produced CPP, indicating the involvement of other β2-containing nAChR antagonist blocked nicotine CPP when infused into the nucleus accumbens (NAc), confirming the critical role of α6β2* nAChRs in this brain region[67]. Similarly, α4 KO mice failed to exhibit nicotine CPP at any dose, suggesting that α4α6β2* nAChRs are necessary for nicotine reward[67].
For cocaine, CPP was abolished in α6 KO mice but preserved in α4 KO mice, implicating α6β2* nAChRs as the primary mediators of cocaine reward[67]. Intra-NAc infusion of MII[H9A; L15A] partially reduced cocaine CPP, suggesting additional contributions from other brain regions or substrates[67]. Control experiments demonstrated that α6 KO mice retained normal associative learning, aversive responses to lithium chloride, and preference for palatable food, indicating that the observed effects were specific to drug reward[67]. Dopamine transporter activity was unchanged in α6 KO mice, ruling out alterations in dopamine uptake as a cause for the reduced CPP[67].
Recently, MII[H9A; L15A] was used in understanding the role of α6* nAChRs in the context of Alcohol Use Disorder (AUD) and Tobacco Use Disorder (TUD), focusing on its ability to modulate ethanol (EtOH)-induced effects on the mesolimbic dopamine system[68]. AUD and TUD often co-occur, and both disorders are associated with dysregulation of the mesolimbic dopamine (DA) system, a key pathway for reward and reinforcement[69,70]. α-MII effectively blocked EtOH’s enhancement of GABAergic input to VTA GABA neurons and EtOH-induced inhibition of GABAergic synaptic transmission to cholinergic interneurons (CINs) in the nucleus accumbens (NAc)[68]. These effects suggest that α-MII-sensitive α6* nAChRs mediate EtOH’s action at these synapses. α-MII also abolished EtOH’s concentration-dependent inhibition of evoked DA release in the NAc, confirming the involvement of α6* nAChRs in regulating CIN-mediated DA release[68]. MII[H9A; L15A] blocked EtOH’s enhancement of CIN firing rate and disrupted inhibitor long-term depression (iLTD) at VTA-NAc GABAergic synapses, a plasticity mechanism linked to alcohol dependence. This all revealed α6* nAChRs as a promising therapeutic target for treating AUD and TUD[68,71].
2.3.2 Framework III, CC-C-C-CC
Framework III is a prominent category in the conotoxin classification system, associated with μ-conotoxins. Prominent examples of conotoxins within Framework III include μ-KIIIA and μ-GIIIA (Figure 5).
|
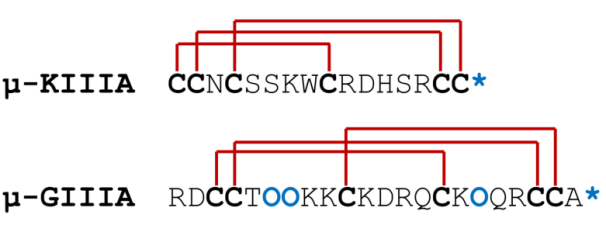
Figure 5. Arrangement of Chosen Framework III Conotoxins for Comparison. Cysteine residues are highlighted in bold black, a star symbol to signify the C-terminus, and O is hydroxyproline. Sequence post-translational modifications are indicated in bold blue.
μ-Conotoxins are distinguished for their targeted inhibition of voltage-gated sodium channels (VGSC), essential in neural and muscular functions[72]. The specific interaction of μ-conotoxins and their VGSC would be of pharmacological interest, not just as drugs themselves, but as blueprints for future medications. This can be seen in Figure 6.
|
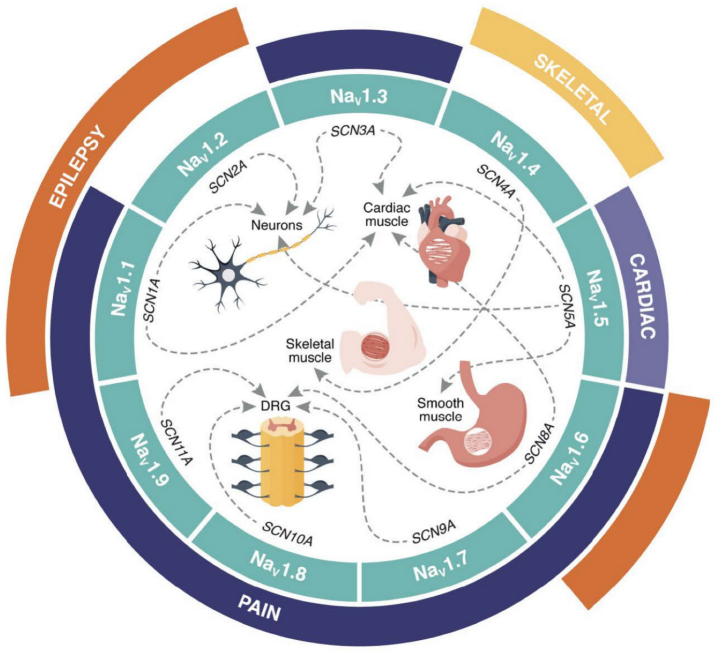
Figure 6. Distribution of Human VGSC Channels and Associated Channelopathies. The nine subtypes (NaV1.1-1.9; teal) are encoded by SCNxA genes (x=1-5, 8-11). Dotted lines show their major tissue expression, while outer segments highlight related channelopathies: pain (dark blue), cardiac (purple), epilepsy (orange), and skeletal muscle disorders (yellow). DRG refers to the dorsal root ganglia. Reproduced from Ref.[75] with permission from MDPI.
2.3.2.1 μ-KIIIA
An example is μ-Conotoxin KIIIA from C. Kinoshita, which primarily targets NaV1.2, a channel integral to transmitting pain signals. μ-KIIIA is among the smaller yet extensively studied μ-conotoxins, particularly noted for its inhibitory action on NaV1.7 channels in humans[76]. This toxin exists in three distinct forms based on disulfide bond configurations: the native form with bonds [C1-C9, C2-C15, C4-C16][77], Isomer 1 with bonds [CI-CV, CII-CIV, CIII-CVI], and Isomer 2 with bonds [CI-CVI, CII-CIV, CIII-CV][78]. Isomer 1 displays varied inhibitory efficacy across NaV subtypes, being most effective against hNaV1.4, then hNaV1.2, and less so on hNaV1.7. In contrast, Isomer 2 inhibits hNaV1.4 and hNaV1.2 equally and shows greater potency than its effect on hNaV1.7. The native form, however, exhibits the most potent inhibition on the skeletal hNaV1.4, with comparable effects on the neuronal hNaV1.7 and hNaV1.2.
In terms of potency, half-maximal inhibitory concentration (IC50) values for Isomer 1 are 124nM for hNaV1.2, 65nM for hNaV1.4, and 413nM for hNaV1.7, showing higher potency compared to the native form, which has IC50 values of 875nM for hNaV1.2, 472nM for hNaV1.4, and 887nM for hNaV1.7[78]. This specificity is highly valuable in researching hNaV1.2 and hNaV1.7 channels and could contribute to developing novel non-opioid analgesics. As well as its interaction with hNaV1.8 recently, although it may not be used in a mutant form as an analog, it’s binding to hNaV1.8 positions it as an excellent pharmaceutical blueprint to study[74,79].
2.3.2.2 μ-GIIIA
Derived from the venom of C. geographus, u-GIIIA, is a pivotal toxin within Framework III and is known for its potent and precise inhibition of NaV channels, a characteristic that makes it noteworthy even among other conotoxins. Its remarkable specificity predominates towards NaV1.4 channels found in skeletal muscle tissues[80]. Recent studies have shown the impressive efficacy of μ-GIIIA. For instance, it has been demonstrated that at a concentration of 10μM, μ-GIIIA can inhibit as much as 99% of NaV1.4 channel activity in the rat models[81]. An IC50 value of 19 nM further highlights this high level of effectiveness[40]. Utilising μ-GIIIA as a research probe offers a promising avenue to explore the pathophysiology of various diseases, especially those related to neuromuscular disorders. These include conditions characterised by paralysis and delayed muscle relaxation, opening potential options for developing novel therapeutic approaches.
In addition, the study of μ-GIIIA holds considerable interest in pharmacological research[82-84]. Its unique ability to selectively target NaV1.4 channels with such potency presents a valuable opportunity in drug development. However, the potential of off-target effects is not to be taken lightly; the biggest concern would be inhibiting all NaV1.4 channels, potentially impacting respiratory muscles. If these muscles were to be paralysed or weakened, it could lead to respiratory failure. This is one of the primary reasons why modulation of NaV1.4 channels has not been explored in drug development due to this risk.
2.3.3 Framework VI/VII C-C-CC-C-C
Framework VI/VII is a classification within the conotoxin family known for encompassing conotoxins with an inhibitory cystine knot (ICK) motif[85]. This framework is vital due to the stability and specificity imparted by the ICK structure, rendering these conotoxins highly effective tools for neurophysiological and pharmacological research. Prominent examples of conotoxins within Framework VI/VII include ω-conotoxin GVIA and ω-conotoxin MVIIC (Figure 7).
|
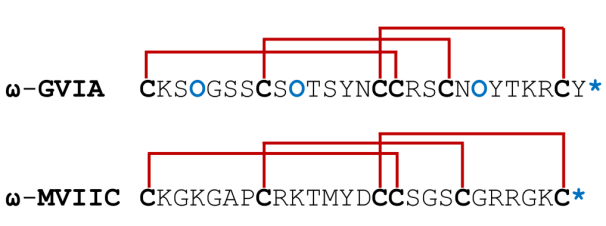
Figure 7. Arrangement of Chosen Framework VI/VII Conotoxins for Comparison. Cysteine residues are highlighted in bold black, a star symbol denotes the C-terminus, and O as 4-hydroxyproline. Sequence post-translational modifications are indicated in bold blue.
2.3.3.1 ω-GVIA
ω-GVIA, sourced from C. geographus venom, is a well-researched peptide in this framework. It irreversibly blocks CaV2.2 channels, which limits its use in potential pharmaceutical applications[86,87]. This potency of ω-GVIA in inhibiting these channels is reflected in its IC50 value of 11.2nM (Figure 8)[36]. Remarkably, studies using a rat model have shown ω-GVIA to exhibit a greater affinity for CaV2.2, with an inhibition constant (Ki) in the picomolar range (2.2pM with 125I-CNVIIA as a competitor and 3.7-4.3pM with 125I-GVIA as a competitor)[88]. The ability of ω-GVIA to block CaV2.2 channels is significant for neuropharmacological studies and its potential therapeutic applications. By inhibiting these channels, ω-GVIA suppresses neurotransmitter release, which plays a crucial role in the modulation of synaptic activity and the overall functioning of the nervous system. Thus, ω-GVIA is an asset in exploring the role of CaV2.2 in synaptic transmission and as a potential blueprint for chronic pain and certain neurological disorders[36]. A drawback is that it lacks the ability to be eluted that ω-MVIIA possesses, which has limited its clinical use.
|
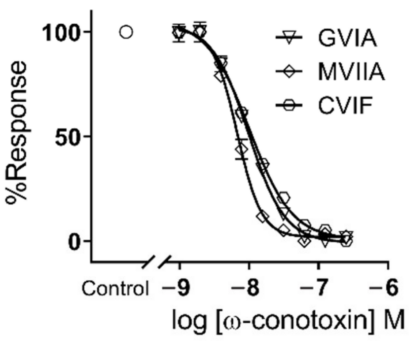
Figure 8. IC50 Dose Response Curve of ω-GVIA. Reproduced from Ref.[36] with permission from MDPI.
2.3.3.2 ω-MVIIC
Isolated from C. magus, exhibits potent and semi-selective inhibition of CaV2.1, with a secondary affinity for CaV2.2[89]. Its structural similarity to ω-MVIIA notwithstanding, ω-MVIIC demonstrates a distinct selectivity profile[90]. ω-Conotoxins such as ω-GVIA and ω-MVIIA, which primarily target CaV2.2, are renowned for their analgesic properties and have been instrumental in research and drug discovery[90,91]. In contrast, ω-conotoxins like MVIIC, which target CaV2.1, are generally not preferred for therapeutic applications. This is attributed to their potential mammalian toxicity and the emergence of adverse effects in clinical settings, casting doubt on their suitability as drug candidates[92]. The crucial role of CaV2.2 in physiological functions, particularly in neurotransmitter release at neuromuscular junctions, further diminishes the appeal of CaV2.1 targeting ω-conotoxins such as ω-MVIIC in pain management. These toxins can be fatal even in small doses. Additionally, in experiments involving a rat model of global ischemia, ω-MVIIC failed to exhibit neuroactive properties[93].
3 CONOTOXINS: A PHARMACOLOGICAL ARSENAL
Currently, conotoxins have been shown to demonstrate beneficial activities against many diseases with numerous classes (as detailed in Figure 2 and Table 1). To maintain focus, this section will primarily address key classes, including α-, ω-, μ-, μO-, μO§, δ-, κ-Conotoxins and two newly identified classes, Con-insulins and Consomatins. While this selection is not exhaustive (other classes include χ-, ε-, γ, ι-, ρ-, σ-, τ-, etc.), it provides a foundation for further detail of recent research. For a deeper dive, we recommend starting with the standard conotoxins of classes not discussed that are listed in Table 1, with synergetic use of the ConoServer database.
3.1 α-Conotoxins
3.1.1 A General Overview of α-Conotoxins
α-Conotoxins, predominately originating from the A superfamily, constitute the most extensively characterised category within the conotoxin domain, primarily acting as antagonists to nAChRs. This interest is due to their targets, nAChRs, which are ligand-gated ion channels, integral to synaptic transmission in both central and peripheral nervous as they bind the neurotransmitter acetylcholine, pivotal in mediating neurotransmission[94-100].
The sequence alignment reveals that the number of amino acid residues separating the 2nd and 3rd cysteine and the 3rd and 4th cysteine varies. The term ‘α-conotoxins framework’ describes the number of residues between these cysteines. For instance, a 4/7 framework indicates four residues between the 2nd and 3rd cysteines and seven residues between the 3rd and 4th cysteines. These intra-cysteine residues are commonly called ‘loops’ within the peptide backbone. The cysteine arrangement results in α-conotoxins adopting a two-loop framework[95]. Figure 9 provides a visual explanation of this concept.
|
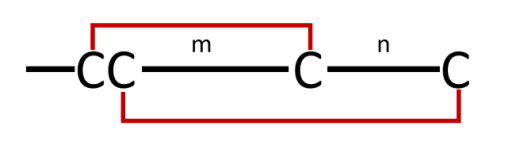
Figure 9. Diagram Illustrating the α-conotoxin Framework. The variables ‘m’ and ‘n’ represent the count of non-cysteine residues located between the 1st and 3rd cysteine residues and the 2nd and 4th cysteine residues, respectively. In identified α-conotoxins, m typically ranges from three to four, while n varies between three and seven.
Due to their extensive range of functions, nAChRs are vital targets in cone snails’ predatory and defensive mechanisms, contributing to the wide variety of nAChR-targeting compounds in their venom. α-Conotoxins block neuromuscular and neuronal nAChR subtypes, both homomeric and heteromeric, with remarkable specificity and effectiveness (see Table 2 for a concise summary of examples). These toxins typically act as competitive antagonists, occupying the acetylcholine binding sites at subunit interfaces (Figure 10). Their specificity is crucial for unravelling the molecular and physiological roles of distinct nAChR variants, as well as for deciphering the physiological roles, precise ligand interactions and selectivity among receptor subtypes[58,59,101-108].
Table 2. Representative Sequences of α-Conotoxins[27]
Name |
Species |
Sequence |
m/n(a) |
Target(s) (IC50) |
Ref. |
Piscivorous |
|
|
Neuromuscular |
|
|
GI |
C. geographus |
|
3/5 |
rα1β1γδ (42nM)> rα1β1δε (339nM)> rα2β2 (5μM)=rα2β4=rα3β2=rα3β4=rα4β2=rα4β4=rα7=rα9 > rα9α10 (9.35μM) |
[31,109-111] |
GIA |
C. geographus |
ECCNPACGRHYSCGK |
3/5 |
α1β1γδ (No IC50 value) |
[30] |
GII |
C. geographus |
ECCHPACGKHFSC* |
3/5 |
α1β1γδ (No IC50 value) |
[30] |
MI |
C. magus |
GRCCHPACGKNYSC* |
3/5 |
mα1β1δ (400pM)> mα1β1γδ (12nM)> rα2β2 (5 μM)=rα2β4=rα3β2=rα3β4=rα4β2=rα4β4=rα7=rα9 >mα1β1;γ (18μM) |
[109,110,112] |
SI |
C. striatus |
ICCNPACGPKYSC* |
3/5 |
rα1β1γδ (113nM)>rα1β1δε (142nM) |
[31] |
SIA |
C. striatus |
YCCHPACGKNFDC* |
3/5 |
α1β1γδ (No IC50 value) |
[113] |
SII |
C. striatus |
GCCCNPACGPNYGCGTSCS |
3/5 |
rα1β1δε (120nM)>rα1β1γδ (370nM) |
[31] |
CnIA |
C. consors |
GRCCHPACGKYYSC* |
3/5 |
mα1β1γδ (190nM)>gα7-5HT3 chimaera (14.8μM) |
[114] |
CnIB |
C. consors |
CCHPACGKYYSC* |
3/5 |
α1β1γδ (No IC50 value) |
[114] |
EI |
C. ermineus |
RDOCCYHPTCNMSNPQIC* |
4/7 |
mα1β1δε (65.9nM)> rα3β4 (6.4μM)> rα3β2 (7.3μM) |
[115,116] |
|
|
|
|
Neuronal |
|
MII |
C. magus |
GCCSNPVCHLEHSNLC* |
4/7 |
rα6/α3β2β3 (390pM)> hα6/α3β4 (1.49nM)> hα3β2 (4.4nM) =ra7*α6β2*> rα4β2 (3.3μM) |
[63,117-119] |
PIA |
C. purpurascens |
RDPCCSNPVCTVHNPQIC* |
4/7 |
rα6/α2β3 (690pM)> hα6/α3β2β3 (1.7nM)> hα6/α3β2β3 (12.6nM)> rα6β4 (33nM)> rα3β2 (74nM)> rα3β4 (518nM)> hα1β1δε (10μM)=rα2β2=rα2β4=rα4β2=rα4β4 |
[120] |
GID |
C. geographus |
IRDγCCSNPACRVNNOHVC |
4/7 |
hα3β2(10nM)=hα7> hα4β2(3μM)> rα4β4(5μM)> hα3β4(10μM) |
[121,122] |
GIC |
C. geographus |
GCCSHPACAGNNQHIC* |
4/7 |
α3β2 (No IC50 value) |
[123] |
AdIA |
C. adamsonii |
GCCSTPPCAVLHC* |
4/4 |
rα6/3 β2β3 (177nM) |
[124] |
|
|
|
Both |
|
|
GaIA |
C. gauguini |
GRCCHPACGRKYNC* |
3/5 |
Musc. rα1β1γδ (38.37nM) Neur. rα3β2 (988.9nM)>rα6/α3β2β3 (1170nM) |
[124] |
Molluscivorous |
|
|
Neuronal |
|
|
PnIA |
C. pennaceus |
GCCSLPPCAANNPD(sTy)C* |
4/7 |
hα7 (510nM)>rα3β2 (300μM) |
[125] |
PnIB |
C. pennaceus |
GCCSLPPCALSNPD(sTy)C* |
4/7 |
α7>α3β2 (No IC50 data) |
[126] |
EpI |
C. episcopatus |
GCCSDPRCNMNNPD(sTy)C* |
4/7 |
rα7 (30nM)>hα3β4 (64.57nM) |
[127,128] |
AuIA |
C. aulicus |
GCCSYPPCFATNSDYC* |
4/7 |
α3β4 (No IC50 data) |
[129] |
AuIB |
C. aulicus |
GCCSYPPCFATNPDC* |
4/6 |
rα3β4 (9.1μM)> mα1β1γδ (75μM) =rα2β2=rα2β4=rα3β2=rα4β4>rα4β2 (300μM)=rα7 |
[129,130] |
AuIC |
C. aulicus |
GCCSYPPCFATNSGYC* |
4/7 |
α3β4 (No IC50 data) |
[130] |
Vermivorous |
|
|
Neuronal |
|
|
VcIA |
C. victoriae |
GCCSDORCNYDHP(Gla)IC* |
4/7 |
rα9α10 (62.9nM) |
[131] |
RgIA |
C. regius |
GCCSDPRCRYRCR |
4/3 |
rα9α10 (2.6nM)> hα9α10 (510nM)> rα7 (3.31μM)> rα6/α3β4 (10μM) =rα6/α3β2β3=rα4β4=rα4β2=rα3β4=rα3β2 =rα2β4=rα2β2> hα1β1δε (16μM) |
[132-136] |
ImI |
C. imperialis |
GCCSDPRCAWRC* |
4/3 |
hα3β2 (40.8nM)> hα7 (497nM)>hα3β4 (3.39μM)> hα1β1δε (10μM) =hα2β2=hα2β4=hα4β2=hα4β4 |
[50,137] |
ImII |
C. imperialis |
GCCSDRRCRWRC* |
4/3 |
hα7 (571nM)>hα1β1δε (1.06μM)>hα3β2 (9.61μM)> hα2β2 (10μM) =hα2β4=hα3β4=hα4β2=hα4β4 |
[137] |
AnIA |
C. anemone |
CCSHPACAANNQD(sTy)C* |
4/7 |
rα3β2 (5.84nM) |
[138] |
AnIB |
C. anemone |
GGCCSHPACAANNQD(sTy)C* |
4/7 |
rα3β2 (280pM)>rα7 (76nM) |
[138] |
Notes: All PTMs marked in bold blue: O, 4-Hydroxyproline; γ, γ-carboxyglutamate; Gla, Gamma carboxylic glutamic acid; *, C-terminal carboxamide; sTy, Sulfotyrosine. Species: h, human; r, rat; ma, T. marmorata; g, G. gallus; m, mussel; (a), α-conotoxin framework; m/n: m and n refer to the number of residues between the 2nd and 3rd Cys and between the 3rd and 4th Cys; Cys residues involved in disulfide bonding are shown in red bold.
|
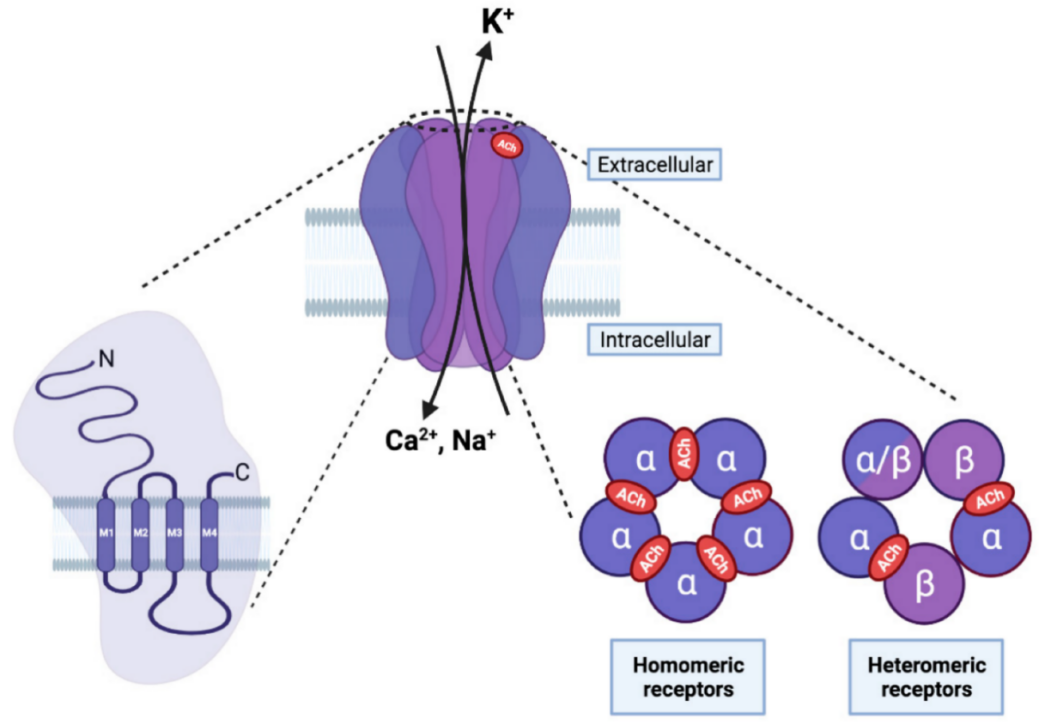
Figure 10. Structure and Function of the nAChR, a Ligand-Gated Ion Channel in the Neuromuscular Junction that Mediates Synaptic Transmission Between Motor Neurons and Muscle Cells. On the left, each nAChR subunit consists of an extracellular amino-terminal region, followed by three hydrophobic transmembrane domains (M1-M3), a large intracellular loop, a fourth transmembrane domain (M4), and an extracellular carboxy-terminal region. In the center, the pentameric arrangement of nAChR subunits is depicted in the fully assembled receptor. The M2 transmembrane domain of the five subunits forms the central pore, critical for selectivity, permeability, and channel gating. On the right, five subunits are assembled, which can create either homopentameric receptors (composed of five alpha subunits, as an example) or heteropentameric receptors (a combination of different subunits). The orthosteric ligand-binding site is located between two alpha subunits (highlighted in red) in homomeric receptors or between the alpha and beta subunits in heteromeric receptors. Reproduced from Ref.[139] with permission from MDPI.
3.1.2 α-GaIA and α-AdIA
A new study focuses on two novel α-conotoxins, α-GaIA and α-AdIA, obtained from the venom of the Polynesian cone snails C. gauguini and C. adamsonii[124]. A one-pot disulfide bond synthesis method was employed, using the 2-nitrobenzyl (NBzl) protecting group for cysteines, to effectively achieve the α-conotoxin fold [Cys 1-3; 2-4]. The investigation of these conotoxins pharmacological characteristics has been made more accessible by advances in chemical synthesis methods and pharmacological characterization[140]. The pharmacological profiling of the synthesised α-conotoxins was assessed, particularly their potency and selectively against various subtypes of rat nAChRs. The findings are quite revealing (which can be seen in Figure 11): α-GaIA showed potent inhibitory activity against neuromuscular nAChR α1β1γδ (IC50=38.37nM). α-GaIA also inhibited the neuronal α3β2 nAChRs and the α6/α3β2β3 nAChR, which are closely related, albeit with about 25-fold less potency (IC50=988.9 nM and IC50=1170nM, respectively). α-AdIA was most effective against the neuronal α6/3 β2β3 subtype of nAChR, displaying an IC50 of 177nM. This research could help develop better ligand inhibition subtypes to understand the functions of nAChR subtypes in physiological and pathological studies. α-AdIA targets both neuronal and neuromuscular nAChRs is notable and significant, as it was previously believed that α-conotoxins would target either neuronal or neuromuscular nAChR subtypes.
|

Figure 11. Dose-Inhibition Relationship for α-AdIA (on the left) and α-GaIA (on the right) Against Specific Subtypes of Rat nAChR, Expressed in Xenopus Laevis Oocytes. Reproduced from Ref.[124] with permission from MDPI.
3.1.3 A General Pre-Clinical Overview of α-Conotoxins
α-Conotoxins exhibit considerable promise in pharmacology, offering diverse applications. These include their use as imaging tools and therapeutic agents for a variety of conditions such as cancer, mental health disorders, cognitive impairments, microbial infections, and the management of neuropathic pain[94].
3.1.3.1 Imaging Tools: α-LvIA, α-LvΙB
As previously discussed in Section 2.3.1.2, conotoxins have served as molecular probes in neurophysiology research for over two decades. Advancements in computational tools and fluorescent labelling techniques have significantly enhanced their utility, enabling precise determination of receptor type concentration and distribution within brain tissue. This has potential future implications for the targeted treatment of various disorders. Initially, α-conotoxins, which can be readily mutated to generate highly potent analogs, have been the primary focus of this research approach.
Fluorescently labelled analogs of the α-conotoxin [Q1G, ∆R14]LvIB, a potent inhibitor (IC50=97nM) of the α7 nAChR, which is implicated in learning, reward, inflammation, and various disorders including Alzheimer’s disease, epilepsy, and schizophrenia, have been used to determine the concentration of α7 nAChR in rat hippocampus, striatum, and cortex[141-145]. The parent peptide [Q1G, ∆R14]LvIB, which carries two disulfide bonds (Cys2-Cys8, Cys3-13) and lacks lysine residues, was folded in vitro through a two-step oxidation. This process was done by (i) (K3[Fe(CN)6]), which facilitated the formation of the first disulfide, followed by (ii) I2-mediated removal of acetamidomethyl (ACM) protection for the second bridge. Reversed-phase HPLC (2-70% buffer B in 30min) and mass spectrometry confirmed successful folding. To create fluorescence analogues, the free N-terminus was conjugated with three NHS-activated dyes: 6-TAMRA-SE, Cy3 NHS ester, and BODIPY-FL NHS ester, resulting in LvIB-R, LvIB-C, and LvIB-B, respectively. The absorption/emission (Ex/Em) profiles closely resembled those of the unmodified dye values: LvIB-R (555/580nm), LvIB-c (550/570nm), and LvIB-B (505/510nm)[145]. Separate electrophysiological experiments on the α7 nAChR and the α3β4 nAChR were able to demonstrate that novel fluorescent conotoxin probes enable the precise valuation of nAChRs and provide the groundwork for more detailed mechanistic studies of their role in addiction, pain and other neurological disorders[145,146].
3.1.3.2 Therapeutic Agents
3.1.3.2.1 Cancer: α-TxID
α-TxID has been used to quantify the expression of various nAChR subunits at both mRNA and protein levels across different lung cancer cell lines compared to normal human embryonic lung fibroblasts (HEL). Using real-time quantitative PCR (qPCR), it was observed that the α3 and β4 subunits were significantly upregulated in the non-small cell lung carcinoma (NSCLC) cell lines A549 and NCI-H1299, with α3 showing approximately 2-fold and 1.8-fold increases, and β4 exhibiting about 1.2-fold and 1.8-fold higher expression respectively compared to HEL cells. Western blot analyses corroborated these findings, demonstrating a 1.3 to 1.7-fold increase in α3 and a 2.1 to 2.2-fold elevation in β4 protein levels in A549 and NCI-H1299 cells relative to HEL controls. Additionally, it was reported that other subunits such as α4, α5, and α7 were also upregulated in certain cell lines, with α5 showing up to an 80-fold increase in A549 cells. These differential expression patterns highlight the potential role of specific nAChR subunits, particularly α3β4, in lung cancer pathogenesis[147].
Building upon these expression data, the therapeutic potential of α-TxID was evaluated. Treatment with α-TxID demonstrated concentration-dependent inhibition of cell proliferation in A549 and NCI-H1299 cells, with significant inhibition observed at higher concentrations (60μM and 120μM). Importantly, when combined, α-TxID exhibited a synergetic effect where the inhibitory rates surpassed those in HEL cells. Specifically, α-TxID alone inhibited approximately 6% of A549 cell proliferation at 0.625μM and up to 92% when combined with an equal concentration of Adriamycin (ADM). Similarly, in NCI-H1299 cells, TxID combined with ADM achieved an inhibitory rate of roughly 70% at 1.25μM concentrations, compared to 41% with ADM alone. This indicates that TxID effectively affects lung cancer cell growth by targeting overexpressed α3β4 nAChRs, inhibiting cell growth and enhancing the efficacy of conventional chemotherapy agents like ADM. The synergistic effect suggested that α-TxID could be integrated into existing treatment regimens to improve therapeutic outcomes for lung cancer patients[147].
3.1.3.2.2 Addiction: α-TxIB
α-TxIB is a promising therapeutic agent for addiction[148]. Utilising a conditioned place preference (CPP) paradigm in C57BL/6J mice, they were administered varying doses of α-TxIB (0, 0.1, 1, and 10nmol) intracerebroventricularly (i.c.v.) before morphine injections (5mg/kg, subcutaneous) during the conditioning phase. This demonstrated that α-TxIB significantly attenuated the expression of morphine-induced CPP at the highest dose of 10nmol/mouse. A lower dose of 1 nmol/mouse showed a non-significant downward trend. During the acquisition phase, pretreatment with 10nmol α-TxIB completely abolished the CPP response α-TxIB did not induce any preference or aversion on its own, as evidenced by the lack of significant change in CPP scores and travelled distance in saline-treated mice[149].
Later, the impact of α-TxIB on learning and memory was assessed using the Morris Water Maze (MWM) and on locomotor and anxiety-like behaviours through locomotor activity tests and the Elevated Plus Maze (EPM). α-TxIB treatment at doses up to 10 nmol/mouse did not impair performance in the MWM, with no significant differences observed in escape latency or spatial memory metrics such as target crossings and time spent in the target quadrant. Similarly, locomotor activity measurements revealed no significant changes in the distance travelled on the first and fourth days of α-TxIB administration. However, a substantial increase in the percentage of distance travelled in the central area was noted at low and medium doses after four consecutive days. The EPM results further confirmed that α-TxIB did not affect anxiety-like behaviours, as no significant changes were observed in the time spent or distance travelled in the open areas. This indicates that α-TxIB effectively inhibits morphine-induced CPP without adversely affecting cognitive functions or inducing anxiety[149].
In alcohol withdrawal studies, zebrafish were exposed to 0.2% alcohol for 14d, followed by 7d of repeated withdrawal. Post-withdrawal, the zebrafish were retro-orbitally injected with 1mg/kg of α-TxIB or saline as a control. Behavioural assessments using the Open Field Test, conducted with 30 zebrafish per group, revealed that the withdrawal group had a significantly reduced max speed of 16.81±1.82cm/s compared to the control group at 23.50±1.52cm/s. α-TxIB administration restored the max speed to 23.67±2.01 cm/s, closely aligning with the control levels. Additionally, global activity in the centre zone decreased from 79.45±16.53cm2/s in controls to 27.03±5.66cm2/s in the withdrawal, with α-TxIB treatment slightly increasing it to 49.58±10.16cm2/s. Monoamine neurotransmitter levels, dopamine, serotonin, noradrenaline, and GABA did not show significant changes after alcohol withdrawal or α-TxIB treatment. However, slight increases were observed in the α-TxIB group compared to controls[150].
Transcriptomic analysis through RNA sequencing identified 657 differentially expressed genes (DEGs) due to alcohol withdrawal, with 344 DEGs differing between the withdrawal and α-TxIB-treated groups. Of these, 225 genes were reversed by α-TxIB injection, primarily enriched in calcium ion binding and calcium signalling pathways. RT-PCR validation confirmed that genes such as myI4 were upregulated 16-fold by withdrawal and normalised by α-TxIB, while atp2a1 was upregulated 9-fold and reduced to 5-fold with α-TxIB treatment. Protein-protein interaction (PPI) network analysis revealed 176 nodes and 621 edges, with an average node degree of 7.06 and a PPI enrichment P<1.0e016. The top 10 hub genes were predominantly involved in muscle contraction processes. This finding indicates that α-TxIB effectively ameliorates behavioural abnormalities induced by alcohol withdrawal in zebrafish by modulating gene expression within the calcium signalling pathway[150].
3.1.3.2.3 Microbial Infection: α-RgIA
α-RgIA was engineered into nine mutant analogues by introducing D-amino acids (D-Lys or D-Arg) and substituting various residues (Gly, Ser, Asp, Pro, Tyr) with Leu or Lys. These changes aimed to boost antimicrobial potential and stability, particularly against four pathogens: Escherichia coli, Bacillus subtilis, Candida tropicalis, and Candida parapsilosis. The minimal inhibitory concentration (MIC) assays showed that certain analogues – especially Pep 6 and Pep 8-displayed potent activity at micromolar levels: Pep 6 and Pep 8 inhibited C. tropicalis down to 8μM, while Pep 8 also inhibited B. subtilis at 16μM. Growth curves demonstrated that even 0.5 x MIC was sufficient to suppress bacterial or fungal growth within a 14h window, and time-kill kinetics revealed a 1,000-fold reduction (bactericidal effect) within just 20-60min at 4 x MIC. Subsequent stability tests in 100% serum, simulated intestinal fluid (SIF), and simulated gastric fluid (SGF) confirmed that D-amino-acid-rich peptides (Pep 5, 6, and 8) resisted proteolysis for up to 24h. In contrast, their L-amino-acid counterparts (Pep 7 and 9) were rapidly degraded within an hour. Further tests at 80℃ showed that these D-containing peptides retained structural integrity for at least 60min[151].
Safety evaluations underscored the relatively low toxicity of several analogues. Hemolysis assays revealed that Pep 5, 6, and 8 induced under 5% hemolysis at 128μM, suggesting minimal damage to red blood cells. While Pep 6 and Pep 9 were nontoxic to human hepatocytes (THLE-3) up to 128μM, Pep 8 caused notable cytotoxicity at ≥64μM, possibly due to stronger electrostatic interactions from its high D-Arg content. In vivo tests in C57BL/6 mice demonstrated that administering Pep 6 at 10mg/kg daily for 7d produced no significant weight loss or overt toxicity, highlighting its promise for future development. Mechanistic imaging using laser confocal microscopy (DAPI/PI staining) and scanning electron microscopy showed cell membrane disruption in both B. subtilis and C. tropicalis following treatment, confirming that membrane permeabilization underlies the antimicrobial action. Leveraging D-amino acid substitutions and strategic inception established a set of RgIA-based antimicrobial peptides with enhanced bactericidal/fungicidal potency, high proteolytic stability, and acceptable safety profiles[151].
3.1.3.3 Neuropathic Pain Management
3.1.3.3.1 α-RgIA-5474
In this section, α-RgIA will be further discussed in neuropathic pain management by examining the efficacy of synthetic RgIA-5474 in alleviating oxaliplatin-induced cold allodynia. RgIA-5474 demonstrated exceptional potency and selectivity by blocking mouse α9α10 nAChRs with an IC50 of 30pM and a Hill slope of -1.0 while exciting over 30,000-fold selectivity compared to α7 nAChRs (IC50 of 1.1μM) and showing minimal inhibition of α4β2 and α3β4 nAChRs at concentrations up to 10μM. In behavioural assays, a single oxaliplatin injection at 10mg/kg for CBA/CaJ mice and 20mg/kg for C57BL/6J mice induced significant cold allodynia, which was fully reversed on day five by administering RgIA-5474 at 40μg/kg/d subcutaneously in wild-type (WT) mice. However, this analgesic effect was absent in α9 knockout (KO) mice, as well as in both male and female mice depleted of CD3* T-cells or with a condition known as the α9 subunit in immune cells using the Cre-loxP system. Quantitative PCR confirmed a significant reduction in chrna9 mRNA levels (-4.8±0.8) in Tie2Cre (+/-) α9loxP (+/+) mice compared to WT (-2.7±0.004) while chimaera 10 levels remained unchanged. This indicates that the α9 nAChR subunit is essential for the analgesic properties of RgIA-5474[152].
3.1.3.3.2 α-PeIA Analogues
Four Dab/Dap-substituted analogues of α-PeIA significantly enhanced potency against human α9α10 nAChRs. Specifically, PeIA[S4Dab], PeIA[S4Dap], PeIA[S9Dap], and PeIA[S4Dap, S9Dap] exhibited IC50 concentrations of 5.32nM, 1.74nM, 4.67nM, and 0.93nM respectively, representing approximately 4-, 13-, 5-, and 24-fold improvements over the native α-PeIA (IC50=21.9nM). Additionally, PeIA[S4Dap, S9Dap] demonstrated high selectivity, remaining inactive (<1% inhibition) at hα1β1εδ, hα1β1δγ, hα3β4, hα4β2, and hα4β4 nAChR subtypes at concentration of 10 and 100 nM. It also inhibited hα3β2 nAChRs with an IC50 of 32.8nM and hα7 nAChRs with an IC50 of 161.7nM, whereas the native α-PeIA was inactive at hα7. Molecular dynamics simulations revealed that the Dap substitutions in PeIA[s4Dap, S9Dap] formed additional hydrogen bonds with receptor residues D166, S168, D169, and Q34, contributing to its enhanced potency. This makes PeIA[S4Dap, S9Dap] one of the most potent and selective α-conotoxin analogues for targeting human α9α10 nAChRs, and positioning is a promising drug candidate for treating chronic neuropathic pain[153].
3.1.3.3.3 α-Mr1.1
A novel conotoxin, α-Mr1.1, exhibited an IC50 value of 92.0nM at hα9α10 nAChRs. Through the development of analogues, the most potent variant, Mr1.1[S4Dap], achieved an IC50 of 4.0nM at hα9α10 nAChRs, representing a>20-fold improvement in potency. Additionally, Mr1.1[S4Dap] demonstrated an IC50 of 20.6nM at the α3β2 subtype and 259.1nM at the α7 subtype, highlighting its high selectivity for α9α10 over other nAChR subtypes. In vivo assessments using a rat chronic constriction injury (CCI) pain model revealed that intravenous administration of Mr1.1[S4Dap] at 25μg/kg sustained a high paw withdrawal threshold (PWT) for up to 24h, significantly outperforming the parent α-Mr1.1 achieving comparable analgesic efficacy to gabapentin at 60mg/kg within the first 7h post-administration. This advancement brought α-Mr1.1 and its analogue Mr1.1[S4Dap] to the forefront as a highly potent and selective candidate for treating chemotherapy-induced neuropathic pain[154].
This was followed up by synthesising 31 analogues of Mr.1[S4Dap] to enhance serum stability and potency against hα9α10 nAChRs. Notably, the analogue Mr1.1[S4Dap, C16Pen] achieved an IC50 of 4.0nM, maintaining potency comparable to the parent compound Mr1.1[S4Dap]. This analogue exhibited a significant increase in serum stability, with 90.93% of the peptide remaining intact after 24h and an extended half-life of 141.8h, compared to 58.60% and 26.2h for Mr1.1[S4Dap], respectively. Selectively assays revealed that Mr1.1[S4Dap, C16Pen] had IC50 values of 4.1nM at hα9α10nAChrs, 21.8nM at hα3β2, and 132.8nM at hα7, demonstrating high specific for α9α10 over other nAChR subtypes. In vivo evaluations showed that intravenous administration of Mr1.1[S4Dap, C16Pen] at doses of 500 μg/kg effectively reduced oxaliplatin-induced cold allodynia for up to 48h, outperforming Mr1.1[S4Dap], which provided relief for only 24h. Additionally, in the cold plate assay, Mr1.1[S4Dap, C16Pen] at 500μg/kg increased the latency to pain reaction to over 140sec compared to 80sec in the control. This has now placed Mr1.1[S4Dap, C16Pen] as a highly potent and stable candidate for chemotherapy-induced neuropathic pain, demonstrating a 5-fold increase in serum stability and maintaining excellent in vitro and in vivo efficacy[155].
3.1.3.3.4 αO-GeX-2
A recently characterized analgesic peptide αO-GeX-2 exhibited an IC50 value of 25.7nM at hα9α10 nAChRs and 18.3nM at rα9α10 nAChRs. αO-GeX-2 also inhibited hα7 nAChRs with an IC50 value of 43.8nM while showing minimal to no inhibition (<5%) at other human nAChR subtypes at 30nM and 100nM concentrations. At GABAB receptor-coupled CaV2.2 channels, αO-GeX-2 demonstrated an IC50 of 4.6nM. In vivo, a single intramuscular dose of αO-GeX-2 at 250μg/kg significantly increased the paw withdrawal threshold (PWT) and paw withdrawal latency (PWL) in a rat chronic constriction injury (CCI) pain model, with analgesic effects lasting up to 24 hours, outperforming morphine administered at 5mg/kg within the first 7h. Additionally, repeated daily intramuscular injections of αO-Gex-2 over 14 days at doses ranging from 0.25μg/kg to 250μg/kg maintained significant analgesic effects without causing motor impairment or addiction, which was proved by conditioned place preference and rotarod tests. This found αO-GeX-2’s potent and selective dual inhibition of α9α10 nAChRs and GABAB-receptor/CaV2.2 channels, promising efficacy and safety profile for treating neuropathic pain[156].
3.2 ω-Conotoxins
ω-Conotoxins, among the first conotoxins characterised, are potent inhibitors of voltage-gated calcium channels (VGCCs). As VGCCs play critical roles in muscle contraction and neurotransmitter release, processes central to pain transmission, development, and maintenance, VGCC inhibitors or modulators present promising therapeutic targets for neuropathic pain[86,157,158],.
Numerous ω-conotoxins have been identified as selective antagonists for specific VGCC subtypes, as seen in Table 3, usually CaV2.1 or CaV2.2. These toxins demonstrated efficacy in preclinical models, effectively reducing allodynia and hyperalgesia in both inflammatory and neuropathic pain conditions[3].
Table 3. Representative Sequences of ω-Conotoxins[27]
Name |
Sequence |
Target(s) (IC50 data) |
Ref. |
Piscivorous |
|
|
|
C. geographus |
|||
GVIA |
|
hCaV2.2 (11.2nM) |
[36] |
GVIIA |
CKSOGTOCSRGMRDCCTSCLLYSNKCRRY |
rCaV2.2 (3.7nM) |
[27,159] |
GVIIB |
CKSOGTOCSRGMRDCCTSCLSYSNKCRRY |
CaV (No IC50 data) |
[27,159] |
C. magnus |
|
|
|
MVIIA |
CKGKGAKCSRLMYDCCTGSCRSGKC* |
hCaV2.2 (6.8nM) |
[36] |
MVIIB |
CKGKGASCHRTSYDCCTGSCNRGKC* |
rCaV2.2 (101pM) |
[27,159] |
MVIIC |
CKGKGAPCRKTMYDCCSGSCGRRGKC* |
^rCaV2.1 (0.6nM)>rCaV2.2 (7.0nM) |
[89] |
MVIID |
CQGRGASCRKTMYNCCSGSCNRGRC* |
CaV2.2 (No IC50 data) |
[160] |
C. striatus |
|
|
|
SVIA |
CRSSGSOCGVTSICCGRCYRGKCT* |
rCaV2.2 (1.46μM) |
[27,159] |
SVIB |
CKLKGQSCRKTSYDCCSGSCGRSGKC* |
rCaV2.2 (1.09nM) |
[27,159] |
SO3 |
CKAAGKPCSRIAYNCCTGSCRSGKC* |
hCaV2.2 (160nM)>rNaV TTX-R (100μM) |
[161,162] |
C. tulipa |
|
|
|
TVIA |
CLSOGSSCSOTSYNCCRSCNOYSRKC |
rCaV2.2 (228pM) |
[27,159] |
C. radiatus |
|
|
|
RVIA |
CKPOGSOCRVSSYNCCSSCKSYNKKCG |
rCaV2.2 (893pM) |
[27,159] |
C. catus |
|
|
|
CVIA |
CKSTGASCRRTSYDCCTGSCRSGRC* |
rCaV2.2 (0.56nM)>rCaV2.1 (850nM) |
[89] |
CVIB |
CKGKGASCRKTMYDCCRGSCRSGRC* |
rCaV2.2 (12nM) |
[163] |
CVIC |
CKGKGQSCSKLMYDCCTGSCSRRGKC* |
^rCaV2.2 (7.6nM)>rCaV2.1(31nM) |
[89] |
CVID |
CKSKGAKCSKLMYDCCSGSCSGTVGRC* |
^rCaV2.2 (0.04nM)>rCaV2.1(55μM) |
[89,164] |
CVIE |
CKGKGASCRRTSYDCCTGSCRSGRC* |
rCaV2.2 (11.1nM)> rCaV1.2 (3μM)=rCaV1.3=rCaV2.3 |
[163,165] |
CVIF |
CKGKGASCRRTSYDCCTGSCRLGRC* |
hCaV2.2 (10nM)> rCaV1.2 (3μM)=rCaV1.3=rCaV2.3 |
[36,163] |
C. consors |
|
|
|
CnVIIA |
CKGKGAOCTRL(Mox)YDCCHGSCSSSKGRC* |
^rCaV2.2 (1.13-3.7pM)>rCaV2.1 (179nM) |
[88] |
Molluscivorous |
|
|
|
C. pennaceus |
|||
PnVIA |
GCLEVDYFCGIPFANNGLCCSGNCVFVCTPQ |
L-type VGCCs |
[166] |
PnVIB |
DDDCEPPGNFCGMIKIGPPCCSGWCFFACA |
L-type VGCCs |
[166] |
C. textile |
|
|
|
TxVII |
CKQADEPCDVFSLDCCTGICLGVCMW |
L-type VGCCs |
[167] |
Vermivorous |
|
|
|
C. moncuri |
|||
MoVIA |
CKPOGSKCSOSMRDCCTTCISYTKRCRKYYN |
hCaV2.2 (330nM) |
[168] |
MoVIB |
CKPOGSKCSOSMRDCCTTCISYTKRCRKYY |
hCaV2.2 (600nM) |
[168] |
C. regularis |
|
|
|
RsXXVIA |
CKGQSCSSCSTKEFCLSKGSRLMYDCCTGSCCGVKTAGVT |
rCaV2.2 (2.8μM) |
[169] |
Notes: PTMs in bold blue: O, 4-Hydroxyproline; *, C-terminal carboxamide; Mox, Oxomethionine; Species: h, human; r, rat; Cys residues involved in disulfide bonding are shown in red bold. Data test: ^, Ki.
ω-Conotoxins exhibit a characteristic sequence homology marked by many basic residues, which imparts a net positive charge. Their structure includes a distinctive cysteine knot motif created by three disulfide bonds. This motif forms a ring with the first two bonds and a penetrating third bond, resulting in four loops. These loops vary in sequence, especially in terms of positively charged and hydroxyl-bearing residues, with a consistent presence of a glycine residue in the first loop. Structurally, they typically feature a triple-stranded beta-sheet connected through several turns and display variability in the orientation of the second and fourth loops[170,171].
The most well-known ω-conotoxin, ω-MVIIA, derived from C. magus, is FDA-approved for treating severe chronic pain in patients unresponsive to opioids. However, its clinical utility is significantly limited by sometimes severe neurological and psychiatric side effects in a minority of cases, which necessitate careful doing to maintain an acceptable therapeutic index as well as intrathecal administration[15,16].
3.2.1 ω-CVID (Leconotide)
ω-CVID, marketed as Leconotide (Figure 12), provides an alternative to Prialt®, derived from C. catus, with improved selectivity for CaV2.2 and a reduced side effect profile. ω-CVID’s Ki evidence reported Ki values for rat CaV2.2 channels ranging from between 0.04-0.07nM[89,164]. Like Ziconotide, Leconotide shares a similar structural framework and exerts analgesic effects by blocking CaV2.2 channels. However, its lower toxicity profile allows for intravenous administration, unlike Ziconotide, which necessitates intrathecal delivery.
|
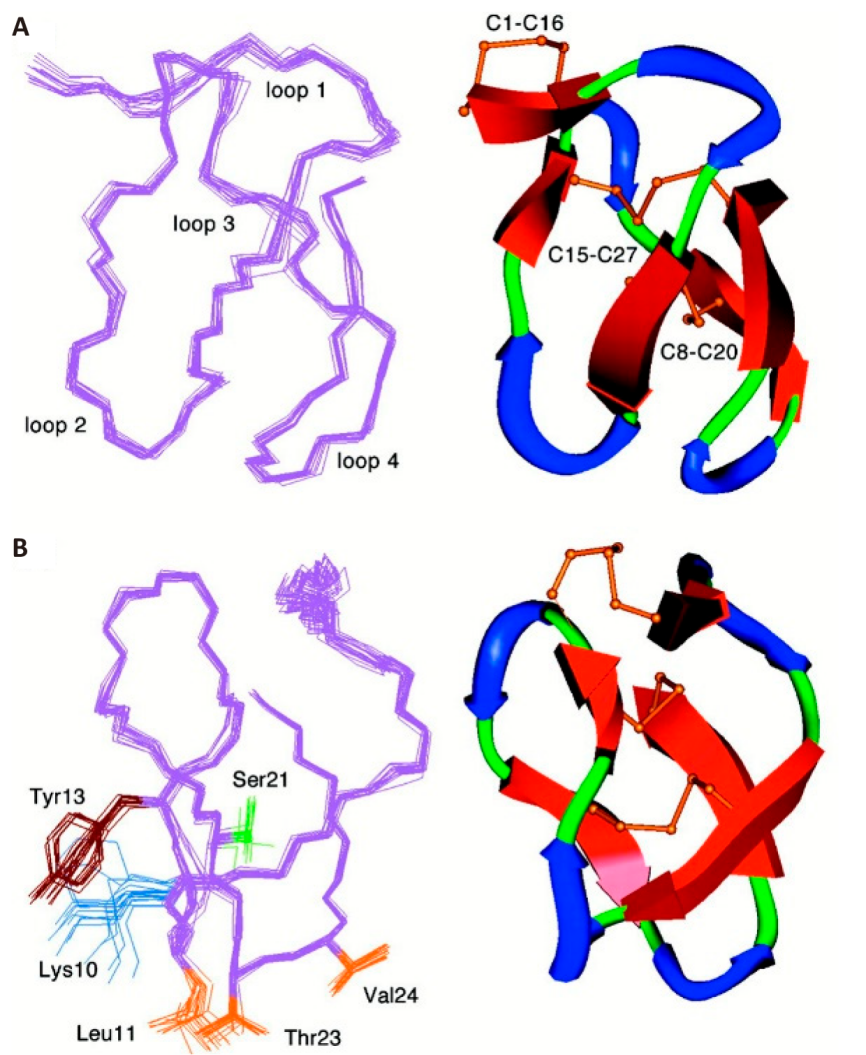
Figure 12. Three-Dimensional Structure of ω-CVID with an Analysis of Its Lowest Energy Conformations. (A) This visualisation presents the superimposition of 20 structures, focusing on the complete backbone and the distinct loops 1 to 4. One specific structure is highlighted to underscore critical features such as beta-bridges and sheets, turns, and the arrangement of disulfide bridges. (B) Additionally, a rotated view reveals the positioning of side chains in loops 2 and 4, contributing to ω-CVID’s activity at CaV2.2, including a novel interaction with Valine. Reproduced from Ref.[89] with permission from Elsevier Inc.
In Phase I/IIa clinical trials, syngeneic rat prostate cancer cells were injected into the tibia of male Wistar rats, inducing bone tumour-related hyperalgesia. The study evaluated the analgesic effects of morphine, Leconotide, and their combination using hot plate assays to measure paw withdrawal thresholds. Leconotide was administered intravenously (IV) alone and in combination with morphine administered intraperitoneally (IP) in alleviating bone cancer-induced hyperalgesia in male Wistar rats. Morphine alone demonstrated a dose-dependent antihyperalgesic effect with an effective 50% (ED50) dose of 2.40±1.24mg/kg IP. When co-administered with Leconotide at 20mg/kg IV, the ED50 of morphine significantly decreased to 0.16±130mg/kg IP, reprising a 15-fold reduction. Additionally, lower doses of Leconotide (0.2mg/kg IV and 0.02mg/kg IV) reduced the ED50 of morphine to 0.39±1.27mg/kg IP and 1.24±1.30mg/kg IP, corresponding to sixfold and twofold reductions, respectively. Leconotide alone, even at the highest non-sedating dose of 20mg/kg IV, did not achieve a 50% reversal of hyperalgesia, demonstrating only a 10.31±3.61% reversal compared to saline controls, which was not statistically significant[172].
Behaviour assessments using the open field activity monitor revealed that high doses of morphine (20mg/kg IP) and Leconotide (200mg/kg IV) induced significant sedation, evidenced by increased low locomotor activity (LMA) of 937.4±22.9sec and 886.1±25.0sec respectively. In contrast, non-sedating doses, including morphine 5mg/kg IP and Leconotide 20mg/kg IV, showed no significant alteration in LMA (P> 0.05). The rotarod test further supported these findings, where only morphine 10mg/kg IP stingingly impaired motor coordination, reducing run time to 108.9±2.3sec. In contrast, morphine 5mg/kg IP did not exhibit a significant effect (118.7±0.2 seconds)[172].
Cardiovascular measurements indicated no major changes in systolic or diastolic blood pressure or heart rate across all treatment groups, including high-dose Leconotide and morphine combinations, ensuring cardiovascular stability. Nociception testing using paw withdrawal latency (PWL) demonstrated that PBS controls had a mean PWL of 12.75sec, while cancer-treated rats exhibited a reduced PWL of 8.1sec, confirming the presence of hyperalgesia. Saline controls showed a 9.3±2.19% reversal of hyperalgesia. Dose-response analysis revealed that coadministration of Leconotide with morphine significantly enhanced the antihyperalgesic effects of morphine. Specifically, the combination of morphine 2.5mg/kg IP with Leconotide 20mg/kg IV (N=5) resulted in a 94.3±3.31% reversal of hyperalgesia, morphine 1.25mg/kg IP with Leconotide 20mg/kg IV achieved 77.8±7.81% reversal, and morphine 5mg/kg IP with Leconotide 0.02 mg/kg IV resulted in 88.7±8.15% reversal. These combinations not only provided superior antihyperalgesic effects compared to either drug alone but also allowed for singingly lower, non-sedating doses of morphine to be used, thereby minimizing opioid-related side effects[172]. This synergistic interaction between Leconotide and morphine holds potential clinical significance, as illustrated in Figure 13.
|
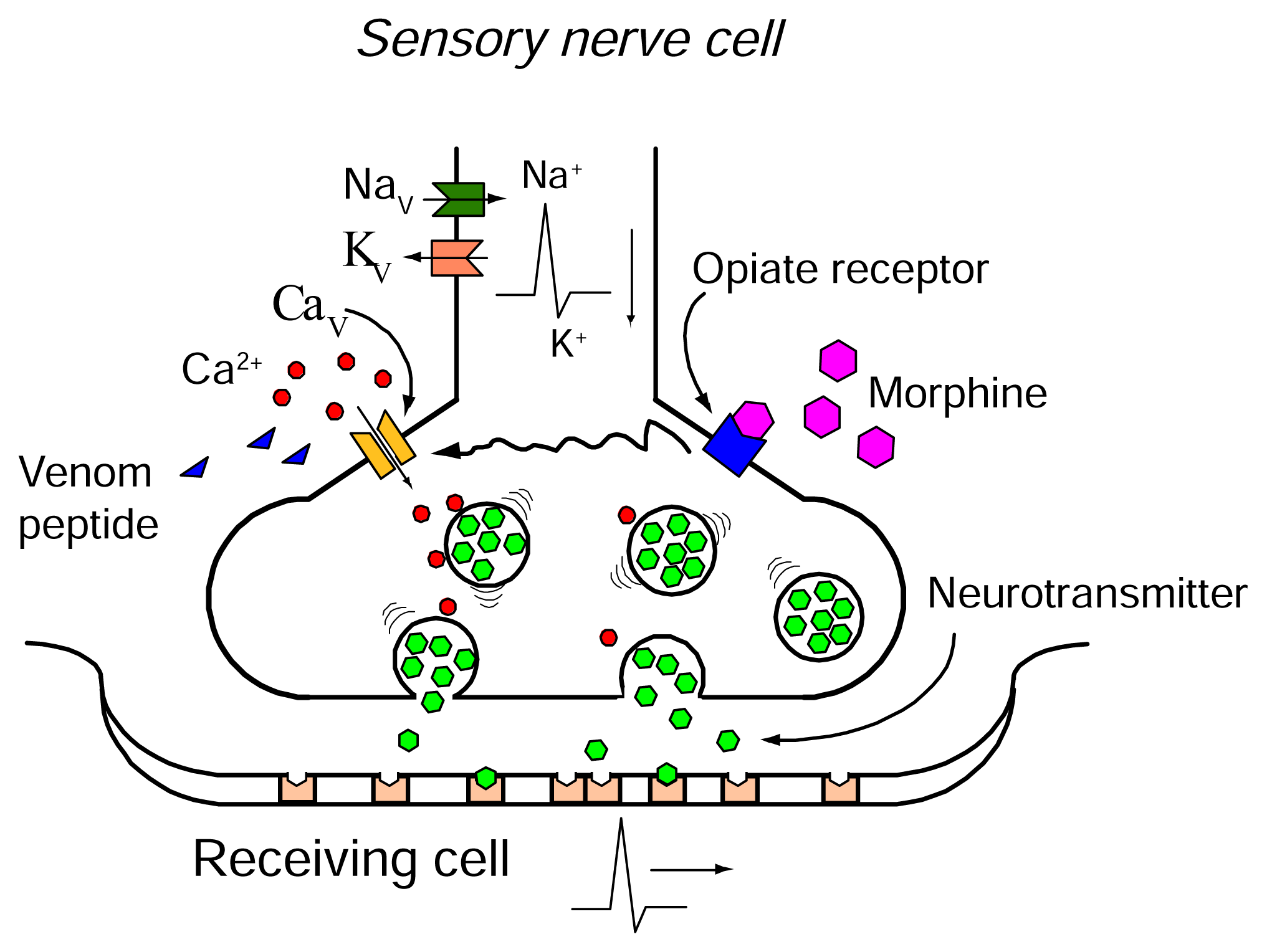
Figure 13. Within Presynaptic Nerve Terminals, the Influx of Calcium Ions Through CaV Channels Is a Crucial Step in Triggering the Release of Neurotransmitters, Thereby Facilitating the Transmission of Pain Signals. Venom peptides such as Prialt® (Ziconotide) and Leconotide can effectively disrupt this process. These peptides effectively interrupt pain signal transmission by blocking the propagation of action potentials and inhibiting calcium influx through CaV channels. Furthermore, the activation of opioid receptors, such as those targeted by morphine, can also modulate CaV channel activity. This occurs through G-protein-coupled receptor mechanisms that alter channel gating and ion permeation. While morphine is often used in combination therapy with Prialt®, Leconotide presents a potentially more advantageous option due to its ability to be administrated intravenously and its generally lower side effect profile compared to Prialt® (Reproduced from Ref.90] with permission from MDPI).
The potential for Leconotide and intravenous administration of conotoxins to enhance the quality of analgesia in patients warrants further investigation. Leconotide’s intravenous administration in combination with morphine offers a more accessible treatment option for a broader patient population, including those ineligible for intrathecal drug delivery via pain pumps[172]. While also suitable for intrathecal administration, clinical trials for CVID have been paused at Phase II due to limited commercial interest at the time for intrathecal therapies[173].
This was coupled in 2012 with a pre-clinical intrathecal administration study in a rat model of neuropathic pain induced by partial sciatic nerve ligation, which demonstrated the analgesic efficacy of both ω-CVID and ω-MVIIA. However, ω-CVID exhibited a marginally better therapeutic index (TI) than ω-MVIIA (1.8-2.4 for ω-CVID vs. 0.7-1.0 for ω-MVIIA)[174].
Hybrid peptides, such as ([K10R]CVID and[R10K]MVIIA, were designed to modify binding properties and reduce motor side effects while maintaining comparable analgesic efficacy. These hybrid peptides demonstrated reduced motor side effects compared to their parent peptides[174].
The degree of inhibition correlated with the recovery of synaptic function after washout, but the recovery time was largely independent of the specific conotoxin used. While ω-CVID and [K10R]CVID exhibited quicker reversibility, ω-MVIIA and [R10K]MVIIA provided longer-lasting effects. These findings challenged the notion of ω-CVID’s superior TI over ω-MVIIA, suggesting that ω-MVIIA might offer longer-lasting analgesia[174].
The development of hybrid mutant peptides, incorporating the beneficial qualities of both ω-CVID and ω-MVIIA, presents a promising avenue for creating more potent and safer therapies with reduced motor side effects[174].
3.2.2 ω-Bu8
ω-Bu8, a novel ω-conotoxin isolated from C. bullatus, is a potent and selective inhibitor of rat CaV2.2 channels, demonstrating significant potential for the treatment of chronic pain[175]. With an IC50 of 89nM, ω-Bu8 exhibits approximately twice the potency of ω-MVIIA against rat CaV2.2[175]. Notably, ω-Bu8 selectively targets CaV2.2 channels with minimal effect on other VGCCs (CaV2.1, IC50 of>10μM and CaV1.1, and minimal inhibition at>10μM) or ion channels (sodium and potassium), highlighting its specificity[17].
In preclinical mouse models of pain, hot plate and acetic acid writhing tests, ω-Bu8 demonstrated potent analgesic effects. At low and moderate doses, ω-Bu8 outperformed ω-MVIIA while achieving comparable efficacy at higher doses. Importantly, ω-Bu8 exhibited significantly fewer side effects on motor coordination, as evidenced by superior performance in the rotarod test. Furthermore, ω-Bu8 demonstrated lower toxicity in goldfish, exhibiting a higher LD50 compared to ω-MVIIA (0.31 mg/kg and 0.21 mg/kg)[175]. These findings suggest that ω-Bu8’s faster binding kinetics (2.33m) and higher recovery ratio (52.6%) compared to ω-MVIIA (3.5m and 42.7%, respectively) contribute to its reduced side effects and faster dissociation from CaV2.2[175].
Structurally, ω-Bu8 possesses the canonical inhibitor cystine knot motif but with a unique loop 2 composition (Arg9 and Thr11), likely contributing to its enhanced potency and selectivity[176,177]. Mutant studies confirmed the critical functional role of these residues, with Bu8[T11A] and Bu8[R9A] having the largest IC50 values (793nM and 418nM, respectively). Moreover, its hydrophilic and flexible loops distinguish ω- Bu8 from ω-MVIIA.
ω-Bu8 is emerging as a promising next-generation CaV2.2 inhibitor, offering a compelling combination of superior efficacy, faster kinetics, and reduced side effects compared to existing therapies. Its characterisation provides valuable insights into the potential for the design of improved analgesics targeting CaV2.2 channels.
3.3 Con-insulins
Many fish-hunting cone snails employ specialised con-insulins in their predatory arsenal (Table 4), with initially identified insulin-like peptides discovered being Con-Ins G1 and Con-Ins G2, derived from the venom of C. geographus[178]. Con-Ins G1 is the smallest naturally derived insulin analogue ever characterised[3]. Also, Con-Ins G1 has evolved adaptations for predation, characterised by its capacity to significantly lower the blood sugar levels of its prey, thus facilitating their capture and immobilisation by inducing a state of hypoglycaemic shock[178].
Table 4. Representative Sequences of Con-insulins[27]
Name |
Sequence |
Target(s) (IC50 data) |
Ref. |
Piscivorous |
|
|
|
C. geographus |
|||
Con-Ins G1 |
|
hIR (11nM) |
[179] |
Con-Ins G2 |
TGYKGIACECCQHYCTDQEFINYCPPVTESSSSSSSAA HEHTCQLDDPAHPQGKCGSDLVNYHEEKCEEEEA |
No IC50 value |
[178] |
Con-Ins G3 |
GIVEVCCDNPCTVATLRTFCH NSDTPKHRCGSELADQYVQLCH |
hIR (46.8nM) |
[179] |
C. floridulus |
|
|
|
Con-Ins F1 |
GIVCECCKNHCNIEELTEYCPPVTEGSG TCESDASPHPQGVCGSPLAEAVEAACEL |
No IC50 value |
[178] |
Con-Ins F2 |
GIVCECCKNHCNIEELTEYCPPVTEGSG TCESDASPHPQGVCGSPLAEAVEAACEL |
No IC50 value |
[178] |
C. kinoshitai |
|
|
|
Con-Ins K1 |
GIVEDCCYNDCTDEKLKEYCHTLQ SDSGTTLVRRRLCGSELVTYLGELCLGN |
hIR (124.4nM) |
[179] |
Con-Ins K2 |
VIVGDCCDNYCTDERLKGYCASLLGL SGTTPDRDHSCGGELVDRLVKLCPSN |
hIR (275.2nM) |
[179] |
C. tulipa |
|
|
|
Con-Ins T1 |
GVVEHCCHRPCSNAEFKKFCG RNSDTPKYRCGSEIPNSYIDLCFR |
No IC50 value |
[178] |
Con-Ins T1A |
GVVEHCCHRPCSNAEFKKFC NSDTPKYRCGSEIPNSYIDLCF |
hIR (9.9nM) |
[179] |
Con-Ins T1B |
GVVEHCCYRPCSNAEFKKFC NSDTPKYRCGSDIPNSYMDLCF |
hIR (18.7nM) |
[179] |
Con-Ins T2 |
GVVEHCCKRACSNAEFMQFC NSDTPWNRCGSQITDSYRELCPH |
hIR (31.8nM) |
[179] |
Con-Ins T3 |
GVVEHCCKRACSNAEFMQFCG NSDTPKYRCGSDIPNSYMDLCFR |
No IC50 value |
[178] |
Con-Ins T4 |
GVVEHCCKRACSNAEFMQFCGNS NSDTPKYRCGSDIPNSYMDLCFR |
No IC50 value |
[180] |
Molluscivorous |
|
|
|
C. marmoreus |
|||
Con-Ins Mr1 |
GIVCECCKNHCTDEEFTEYCPHVTESG VCGSNQPNHPNGKCGSKMADYLEEQCEEEEAA |
No IC50 value |
[178] |
C. textile |
|
|
|
Con-Ins Tx1 |
GIVCECCKHHCTKEEFTEYCH VCWLGDPNHPQGICGPQVADIVEIRCEEKEAEQ |
No IC50 value |
[178] |
Vermivorous |
|
|
|
C. imperialis |
|
|
|
Con-Ins Im1 |
GITCECCYNHCSFRELVQYCN TCTLETRMQGAHPQGICGSKLPDIVHTVCQVMGRGYAGGQRQLRK |
No IC50 value |
[178] |
Con-Ins Im2 |
GITCECCFNQCTYYELLQYCN TCTLATRSRGAHPSGICGRNLARIVSVLCTPRGYVSNWFTKR |
No IC50 value |
[178] |
C. memiae |
|
|
|
Con-Ins Me1 |
GMSCECCKNSCDAEEILEYCPPLPSS TCSPSEPAAPGGICGSNLAELHSFLCEK |
No IC50 value |
[178] |
C. quercinus |
|
|
|
Con-Ins Q1 |
GIVCECCKNSCTYEEITEYCPPVTEGSG TCEPGASPHPQGKCRPELAEFHETMCEV |
No IC50 value |
[178] |
Notes: Species: h, human; Cys residues involved in disulfide bonding are shown in red bold. Disulfide linkages are diverse for Con-insulins, Con-Ins G1 linkage is provided as an example.
Con-insulins are expressed near the venom duct's injection site, attaining a significantly high concentration[181]. In experimental studies, administering Con-Ins G1 to zebrafish through intraperitoneal injection reduced their blood glucose levels effectively, mirroring the impact of human insulin[178,181]. Additionally, when Con-Ins G1 was introduced into the water, there was a notable decrease in the swimming activity and frequency of movements of zebrafish larvae[181]. In diabetic mouse models, Con-Ins G1 demonstrated a significantly higher efficacy in reducing blood glucose levels, up to ten times more potent than human insulin[178]. This specific con-insulin is a significant component of the venom in C. geographus, exerting a considerable hypoglycaemic effect on prey, hindering their escape, and making them easier to capture[3,182]. Like other conotoxins, this multifaceted biological activity of con-insulins highlight their potential as pharmacological therapeutic agents in conditions such as diabetes.
The analysis by Guo et al.[182] of con-insulin diversity reveals species-specific variations in the expression of insulin-like peptides in cone snail venom (Figure 14). Different species express a singular form of coninsulin or multiple variants. These con-insulins generally have a conserved N-terminal signal sequence in their amino acid structures. Notably, some con-insulins incorporate a unique propeptide region, unlike human and zebrafish insulins, which aids in protein folding and prevents misfolding or aggregation during secretion[182].
Thirty-eight distinct insulin sequences and one insulin-like peptide from 18 different species of cone snails have been identified[182]. The signal peptide is highly conserved across these sequences. The mature region of insulin, which includes both A, B, and C chains, displays variability and is connected through disulfide bonds, essential for its pharmacological action[183]. Although the mature con-insulin peptide resembles human insulin, there are significant differences. For example, in insulin sequences, cysteine residues, predominately found in the A and B chains, follow a specific pattern that influences the formation of disulfide bonds. Some coninsulin variants exhibit a unique pattern in their A and B chains, resulting in a different number of disulfide bonds than human insulin[184,185].
Interestingly, certain con-insulins, such as Con-Ins-G1, maintain an affinity for the human insulin receptor despite a shorter or partially absent C-terminus in the B chain[186,187]. This suggests that the B chain's C-terminal segment is not critical for binding affinity with the insulin receptor. Moreover, con-insulins do not typically undergo dimerization or hexamerization, which allows for faster reaction kinetics than insulin medications[182,187]. Con-Ins G1 showed a high affinity for the human insulin receptor and structural similarity to zebrafish insulin[179,181]. This similarity, especially in the A-chain, aligns with the rapid predator strategies of cone snails. Homologous modelling methods were used to generate various con-insulins variants. A structural comparison of insulin molecules reveals three α-helices and a hydrophobic core, crucial for structural integrity and normal folding[182]. Con-insulins from fish-hunting cone snails are like human and zebrafish insulins, attributed to their dietary habits and the need to act on fish insulin receptors. This is attributed to the shorter B-chain in fish-hunting cone snails’ insulin[182].
|
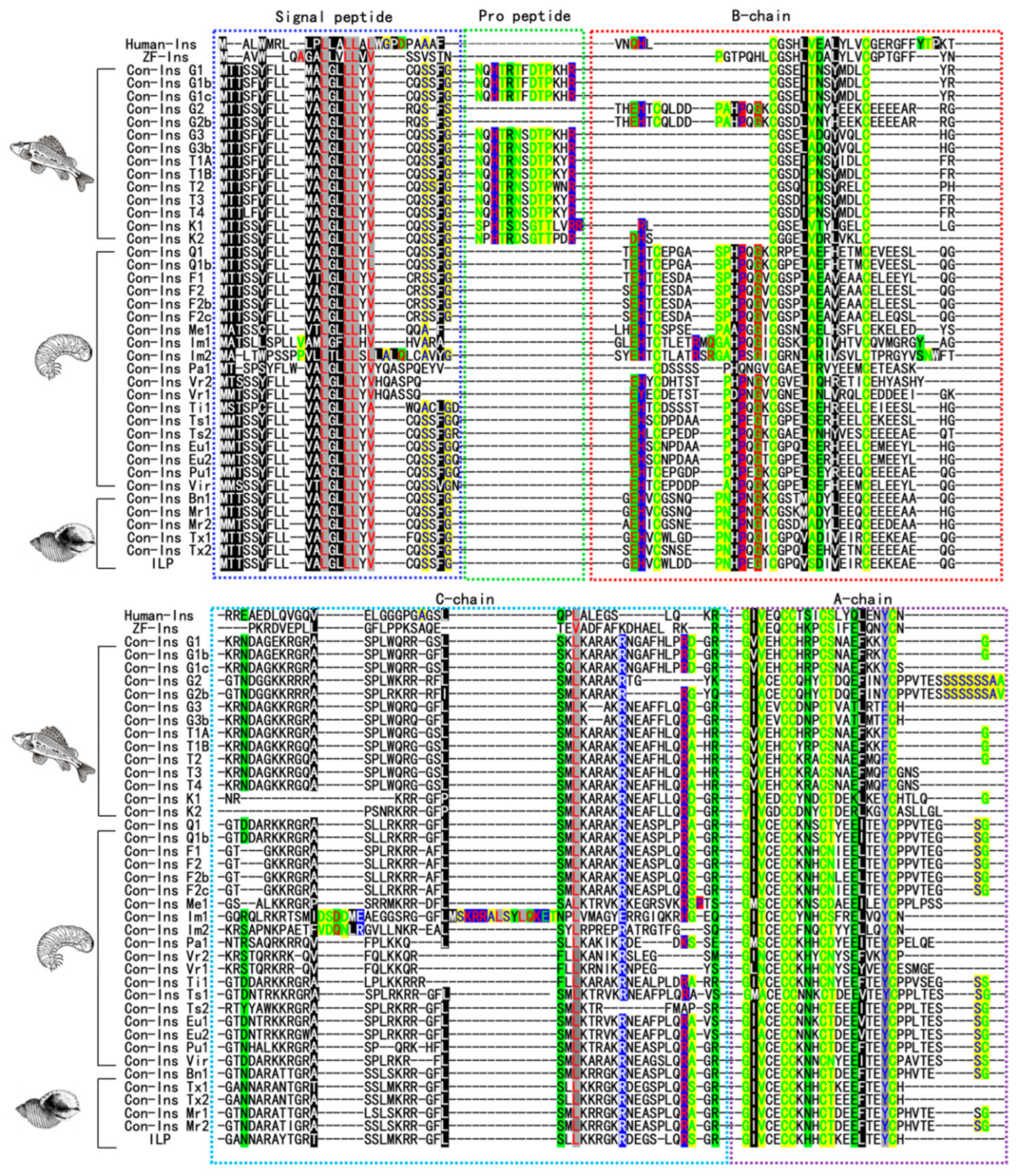
Figure 14. This Sequence Alignment Involved Comparing Insulin Sequences from Humans, Zebrafish, and Various Conus Species. In these alignments, the A-chains and B-chains of insulin are marked in purple and red boxes, respectively. Key features such as conserved cysteine residues and smaller amino acids are highlighted. The alignment also delineates different types of amino acids based on their properties and charge, with distinct colour coding for ease of identification. This includes aliphatic and aromatic amino acids, amphoteric and polar groups, negatively and positively charged amino acids, amphoteric and polar groups, and charged amino acids, each with its colour scheme. Reproduced from Ref.[182] with permission from MDPI.
3.4 µ-Conotoxins
3.4.1 µ-Conotoxins
µ-Conotoxins belong to the M-superfamily of conotoxins and are distinguished as the earliest identified peptide inhibitors of voltage-gated sodium channels (VGSCs)[75,170]. µ-Conotoxins target a specific site, site I, at the extracellular end of the sodium channel pore[188]. µ-Conotoxins display a higher degree of subtype specificity in their interaction with sodium channels than guanidinium toxins such as tetrodotoxin (TTX), being able to distinguish between cardiac, neuronal and skeletal muscle isoforms[189]. µ-Conotoxins are characterised by their sequence homology and abundance of positively charged residues (Table 5). Structurally, µ-conotoxins are compact, featuring multiple turns and helices, stabilised by three disulfide bonds, resulting in a conformation that includes a 310-helix and a beta-hairpin turn[170]. The arginine residue, a crucial element in their structure, plays a vital role in their activity. However, arginine’s interaction with VGSCs in µ-conotoxins differs from that of the guanidinium group in TTX[170,190].
Table 5. Representative Sequences of µ-Conotoxins[27]
Name |
Sequence |
Target(s) (IC50) |
Ref. |
Piscivorous |
|
|
|
C. consors |
|||
CnIIIA |
|
rNaV1.2 (250nM)> rNaV1.4 (270nM)> mNaV1.6 (7.1μM)> rNaV1.5 (7.4μM)> rNaV1.7 (10μM)=rNaV1.8> rNaV1.3 (11μM)> rNaV1.1 (14.2μM) |
[40] |
CnIIIB |
ZGCCGEPNLCFTRWCRNNARCCRQQ |
pNaV TTX-R (10μM) |
[191] |
CnIIIC |
ZGCCNGPKGCSSKWCRDHARCC* |
rNaV1.4 (1.3nM)> rα3β2 (450nM)> rNaV1.2 (500nM) |
[192] |
C. kinoshitai |
|
|
|
KIIIA |
CCNCSSKWCRDHSRCC* |
hNaV1.4 (472nM)> hNaV1.2 (875nM)> hNaV1.7 (887nM)> hNaV1.5 (10μM)=hNaV1.8 |
[78,79] |
KIIIB |
NGCCNCSSKWCRDHSRCC* |
rNaV1.2 (26nM) |
[77] |
C. geographus |
|
|
|
GIIIA |
RDCCTOOKKCKDRQCKOQRCCA* |
rNaV1.4 (69nM)> rNaV1.5 (680nM)> rNaV1.3(10μM)=rNaV1.7=rNaV1.8> rNaV1.2 (17.8μM) |
[40,80] |
GIIIB |
RDCCTOORKCKDRRCKOMKCCA* |
NaV TTX-S (No IC50 Value) |
[193] |
GIIIC |
RDCCTOOKKCKDRRCKOLKCCA* |
rNaV1.4 (286nM) |
[194] |
C. stercusmuscarum |
|
|
|
SmIIIA |
ZRCCNGRRGCSSRWCRDHSRCC |
rNaV1.6 (160nM)> rNaV1.5 (1.3μM)> rNaV1.7 (1.3μM)> rNaV1.8 (10μM) |
[40] |
C. striolatus |
|
|
|
SxIIIA |
RCCTGKKGSCSGRACKNLKCCA* |
rNaV1.4 (7nM)> rNaV1.1 (370nM)> rNaV1.6 (570nM)> rNaV1.2 (1μM)> rNaV1.3 (10μM)=rNaV1.5=rNaV1.7=rNaV1.8 |
[40,195] |
SxIIIB |
ZKCCTGKKGSCSGRACKNLRCCA* |
NaV (Subtypes and IC50 unknown) |
[195] |
SxIIIC |
RGCCNGRGGCSSRWCRDHARCC* |
rNaV1.4 (15.1nM)> rNaV1.3 (89.4nM)> mNaV1.6 (124.9nM)> rNaV1.1 (132nM)> rNaV1.7 (152.2nM)> rNaV1.2 (363.8nM)> rNaV1.5 (5μM)=rNaV1.8 |
[196] |
C. tulipa |
|
|
|
TIIIA |
RHGCCKGOKGCSSRECROQHCC* |
rNaV1.4 (9nM)> rNaV1.2 (40nM)> rNaV1.1 (900nM)> rNaV1.3 (7.9μM)> rNaV1.5 (10μM)=rNaV1.7> rNaV1.5 (25μM) |
[40, 197] |
C. magnus |
|
|
|
MIIIA |
ZGCCNVPNGCSGRWCRDHAQCC* |
rNaV1.5 (10μM)=rNaV1.7> mNaV1.6 (21.6μM)> rNaV1.1 (22.6μM)> rNaV1.7 (97μM) |
[40] |
C. striatus |
|
|
|
SIIIA |
ZNCCNGGCSSKWCRDHARCC* |
rNaV1.4 (560nM)> mNaV1.6 (760nM)> rNaV1.2 (790nM)> rNaV1.3 (5.3μM)> rNaV1.8 (10μM)> rNaV1.1 (11μM)> rNaV1.7 (65μM)> rNaV1.5 (251μM) |
[40,198-200] |
SIIIB |
ZKCCTGKKGSCSGRACKNLRCCA* |
^rNaV1.4 (3.24nM)>rNaV1.2 5.13nM) |
[199] |
C. catus |
|
|
|
CIIIA |
GRCCEGPNGCSSRWCKDHARCC* |
NaV TTX-R (No IC50 data) |
[191] |
C. bullatus |
|
|
|
BuIIIA |
VTDRCCKGKRECGRWCRDHSRCC* |
mNaV1.6 (4.4μM)> rNaV1.7 (10μM)=rNaV1.8> rNaV1.6 (13.8μM) |
[40] |
BuIIIB |
VGERCCKNGKRGCGRWCRDHSRCC* |
rNaV1.5 (9μM)> rNaV1.7 (10μM)=rNaV1.8> mNaV1.6 (30.36μM) |
[40,201] |
Molluscivorous |
|
|
|
C. pennaceus |
|
||
PnIVA |
CCKYGWTCLLGCSPCGC |
NaV TTX-R (No IC50 data) |
[202] |
PnIVB |
CCKYGWTCWLGCSPCGC |
NaV TTX-R + NaV (16nM) |
[202,203] |
Vermivorous |
|
|
|
C. litteratus |
|||
Lt5d |
DCCPAKLLCCNP |
rNaV TTX-R (150nM)> rNav TTX-S (156.16nM) |
[204] |
C. tessulatus |
|
|
|
TsIIIA |
GCCRWPCPSRCGMARCCSS |
hNaV1.8 (2.11μM)> rNaV TTX-R (2.61μM)> hNaV1.1-NaV1.7 (10μM) |
[205,206] |
Notes: PTMs marked in blue bold: Z, pyroglutamate; O, 4-hydroxyproline; *, C-terminal carboxamide; Different test(s) than IC50: ^, Ki; Species: h, human; r, rat; m, mussel; Cys residues involved in disulfide bonding are shown in red bold.
The development of novel pharmacological NaV channel blockers from cone snails as pain therapeutics has been hindered by adverse side effects, such as rapid paralysis and death, due to their off-targeting NaV1.4. This unfavourable outcome has contributed to a decline in the exploration of µ-conotoxins in research and drug development[3,207]. However, recent research has expanded our understanding of µ-conotoxins, particularly their interaction with human VGSC subtypes and engineering mutants to diminish their ability to reduce NaV1.4 off-targeting. For instance, whole-cell patch-clamp electrophysiology has recently been used to assess the potency and selectivity of µ-conotoxins such as µ-SxIIIC, µ-SmIIIA, and µ-KIIIA[207].
3.4.2 µ-SmIIIA and µ-SxIIIC
The mechanisms by which specific µ-conotoxins, notably µ-SxIIIC and µ-SmIIIA, inhibit the human NaV1.7 channel subtype, which plays a critical role in pain perception, is of recent significant interest[207]. Since its discovery, µ-SxIIIC has been recognised for its potent inhibition of NaV1.7[196]. The influence of loop 3’s residue count on this inhibition capacity is critical. Mutagenesis studies revealed that charged residues in loop 3 affect selectivity toward NaV1.4, which is why µ-SxIIIC has enhanced potency against NaV1.7, outperforming µ-KIIIA. These insights, along with the more straightforward structure of µ-SxIIIC to synthesise compared to its counterpart µ-SmIIIA, may assist in developing µ-conotoxins derivatives targeting NaV1.7. However, interestingly, previous optimisation efforts in µ-conotoxins have typically resulted in limited, non-selective NaV1.7 inhibition. Remarkably, it was discovered that µ-SmIIIA, previously believed to weakly inhibit mammalian NaV1.7, is a potent inhibitor of human NaV1.7 (IC50 value of 41±4nM), making it the most effective µ-conotoxin inhibitor of NaV1.7 to date (Figure 15A-B). This emphasises the importance of utilising human cell-cultured receptors in conotoxin research, as the specificity of this conotoxin was significantly underestimated. Intriguingly, substituting an arginine at residue 16 with a histidine in µ-SxIIIC resulted in a threefold improvement in NaV1.7 inhibition (from an IC50 of 152.2±26.8nM to 66.5±2.5nM) compared to wild-type µ-SxIIIC and significantly reduced its inhibition of NaV1.4 sixfold (from an IC50 of 12.8±13.0nM to 80.3±24.7nM) (Figure 15C)[207].
|
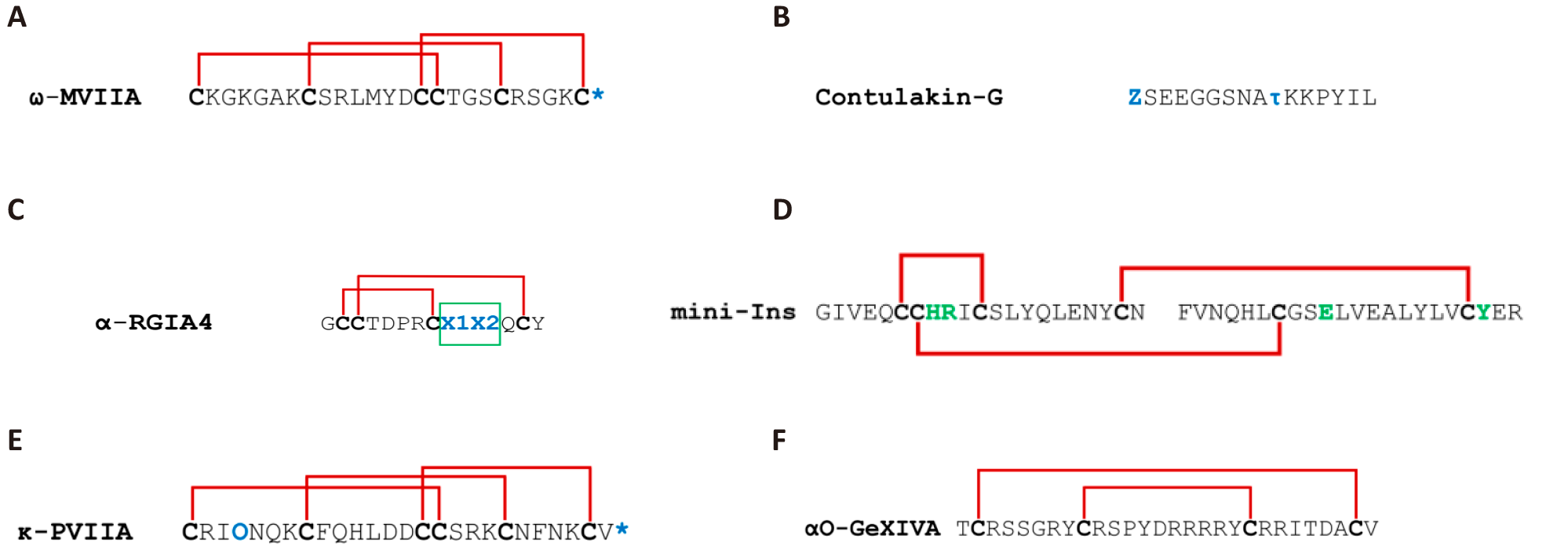
Figure 15. µ-Ctx Dose-response and µ-SxIIIC Mutation Effects on hNaV1.4/hNaV1.7. A: Dose-response curve for hNaV1.4 of µ-SxIIIC (teal), µ-SmIIIA (maroon), and µ-KIIIA (yellow). B: The dose-response curve for hNaV1.7 of µ-SxIIIC (teal), µ-SmIIIA (maroon), and µ-KIIIA (yellow. C: Impact of individual µ-SxIIIC mutations on hNaV1.7 potency and selectivity. pIC50 values±SEM are shown for hNaV1.4 (maroon) and hNaV1.7 (blue). Mutants [R16A]SxIIIC, [D17A]SxIIIC, and [R20A]SxIIIC retained selectivity for hNaV1.4 over hNaV1.7, while mutations ([Δ1,2], [R16H], [R16Q]) reduced at hNaV1.4 without affecting NaV1.7, eliminating subtype selectivity. Data represent mean±SEM (n=3-5), with significance assessed by unpaired t-test (*P<0.05). Reproduced from Ref.[207] with permission from American Chemical Society.
3.5 μO-, μO§, and δ-Conotoxins
3.5.1 μO and μO§ Conotoxins
μO and μO§ conotoxins inhibit NaV channels, while δ-conotoxins act as activators, causing delays in activation (Table 6)[3]. μO-conotoxins, part of the O-superfamily, are hydrophobic and feature three disulfide bonds forming a cystine knot, resulting in four loop structures[170]. Notable peptides from C. marmoreus, μO-MrVIA and μO-MrVIB belong to this group[208]. These toxins selectively inhibit VGSCs at a unique site different from Site I, where μ-conotoxins bind. They modulate voltage sensors in domain II, restricting channel opening rather than blocking the pore[209]. This unique binding site allows μO-conotoxins to avoid competition with saxitoxin and tetrodotoxin and to interact with a broader range of VGSC subtypes. The three-dimensional structure of μO-MrVIB shares a global fold similar to ω-conotoxins[170,210].
Table 6. Representative Sequences of μO-, μO§, and δ-Conotoxins[27]
Class |
Name |
Sequence |
Target(s) (IC50) |
Ref. |
μO |
Molluscivorous |
|
|
|
C. marmoreus |
||||
MrVIA |
ACRKKWEYCIVPIIGFIYCCPGLICGPFVCV |
rNaV1.7 (345nM)> rNaV1.4 (438nM)> rNaV1.2 (532nM) |
[35] |
|
MrVIB |
ACSKKWEYCIVPILGFVYCCPGLICGPFVCV |
rNaV TTX-R (22nM)> rNaV1.8 (326nM)> rNav TTX-S (>1μM) |
[211] |
|
C. magnificus |
||||
MfVIA |
RDCQEKWEYCIVPILGFVYCCPGLICGPFVCV |
hNaV1.4 (81nM)> hNaV1.5 (431nM)> hNaV1.6 (1.2μM)> hNaV1.8 (1.9μM)> hNaV1.3 (2.2 μM)> hNaV1.7 (2.3μM)> hNaV1.1 (3.3μM)> rNaV1.2 (5.1μM) |
[212] |
|
μO§ |
Piscivorous |
|||
C. geographus |
||||
GVIIJ |
GBCGDOGATCGKLRLYCCSGFCD(Scc)YTKTCKDKSSA |
rNaV1.2 (29nM)> rNaV1.7 (32nM) |
[213] |
|
δ |
Molluscivorous |
|||
C. textile |
||||
TxVIA |
WCKQSGEMCNLLDQNCCDGYCIVLVCT |
hCaV3.2 (240nM) |
[214] |
|
TxVIB |
WCKQSGEMCNVLDQNCCDGYCIVFVCT |
NaV (Subtypes and IC50s unknown) |
[208,215] |
|
C. gloriamaris |
||||
GmVIA |
VKPCRKEGQLCDPIFQNCCRGWNCVLFCV |
rNaV1.2 (2.5μM)> rNaV1.4 (4.8μM) |
[35] |
|
Vermivorous |
||||
C. suturatus |
||||
SuVIA |
CAGIGSFCGLPGLVDCCSDRCFIVCLP |
$hNaV1.6 (1.27nM)> hNaV1.7 (2.42nM)> hNaV1.3 (3.98nM)> hNaV1.4 (4.99nM) |
[216] |
|
C. tessulatus |
||||
TsVIA |
CAAFGSFCGLPGLVDCCSGRCFIVCLL |
NaV + KV (Subtypes and IC50s unknown) |
[217] |
|
Piscivorous |
||||
C. aurisiacus |
||||
AVIA |
DGCSNAGAFCGIHOGLCCSEICIVWCT |
NaV (Subtypes and IC50s unknown) |
[218] |
|
C. bullatus |
||||
BVIA |
DECSAOGAFCLIROGLCCSEFCFFACF |
NaV (Subtypes and IC50s unknown) |
[218] |
|
C. purpurascens |
||||
PVIA |
EACYAOGTFCGIKOGLCCSEFCLPGVCFG* |
hNaV1.7 (1.9μM)> rNaV1.2 (2.9μM)> rNaV1.4 (5.2μM) |
[35] |
|
C. striatus |
||||
SVIE |
DGCSSGGTFCGIHOGLCCSEFCFLWCITFID |
rNaV1.4 (500nM) |
[219] |
|
Notes: PTM: Marked in blue bold; B, bromotryptophan; O, 4-hydroxyproline; *, C-terminal carboxamide; Scc, S-cysteinylated cysteine; Tests other than IC50: $, EC50; Species: h, human; r, rat; Cys residues involved in disulfide bonding are shown in red bold.
μO§-GVIIJ, derived from C. geographus, is 35 peptide conotoxin characterised by seven cysteine residues, with six forming three disulfide bonds and the seventh (Cys24) undergoing S-cysteinylation[3,213,220]. This toxin targets site 8 on sodium channels between S5 and SS5 of domain II, creating a disulfide tether with Cys910 of rat NaV1.2 channels. Electrophysiological studies showed that S-glutathionylated μO§-GVIIJ inhibits all TTX-sensitive rat NaV1 subtypes but is not a classical pore blocker[3,213]. Redox conditions influence μO§-GVIIJ’s inhibitory mechanism, as thiol-modifying agents can protect NaV1.2 from inhibition. Research involving μO§-GVIIJ analogues with modified Cys24 has provided insights into the redox state of extracellular cysteines in NaV channels[221]. Additionally, μO§-GVIIJ’s dependency on β-subunits has shown that action potentials in A-fibres of rodent sciatic nerves are primarily mediated by NaV1.6 channels associated with NaVβ2 or NaVβ4 subunits[3,201].
3.5.2 δ-Conotoxins
δ-Conotoxins exhibit a unique mechanism of action by delaying the inactivation of NaV channels, leading to persistent neuronal firing. These toxins differ from µ-conotoxins, which block NaV channels and are more structurally akin to ω-conotoxins, containing three disulfide bonds in a cysteine knot motif[170]. Unlike the relatively hydrophilic ω-conotoxins, δ-conotoxins are inherently hydrophobic, suggesting their binding site on VGSCs may be partially embedded within the lipid bilayer at the S3/S4 linker of domain IV of the NaV channel[3,189]. Initial research focused on δ-conotoxins from piscivorous cone snails such as C. textile, with characterised toxins like δ-TxVIA and δ-TxVIB, which predominately target molluscan NaV[222-224]. δ-NgVIA from C. textile also affected molluscan and vertebrate channels, indicating a broader activity spectrum[225].
Recent studies have identified potent activators of vertebrate NaV channels from worm-hunting cone snails, like δ-TsVIA (C. tessulatus), δ-ErVIA (C. eburneus), and δ-SuVIA (C. suturatus), with δ-SuVIA notably activating human at EC50 NaV1.3 (3.98nM), NaV1.6 (1.27nM), and NaV1.7 channels (2.42nM) at low nanomolar concentrations[3,216,217]. This presence of vertebrate-active δ-conotoxins in nonpiscivorous species suggests an evolutionary adaptation of these toxins, possibly originating as a defence mechanism against larger predators and later repurposed for facilitating a dietary shift in cone snail lineages[3,216]. Despite the rich diversity and unique pharmacological profiles of δ-conotoxins, their therapeutic application remains unexplored, primarily due to challenges in synthesis and purification arising from their hydrophobic nature[3]. Recent advancements using an acid-cleavable solubility tag approach have begun to address these challenges, opening new avenues for exploring the potential of δ-conotoxins in pharmacological applications[226].
3.6 κ-Conotoxins
κ-Conotoxins are renowned for their unique structural architecture, characterized by three disulfide bonds in a 1-3, 2-5, and 4-6 connectivity (Table 7), forming a cystine knot topology. The first κ-conotoxin identified, κO-PVIIA from C. purpurascens, is a pivotal model for understanding potassium channel function. κO-PVIIA potently blocks voltage-gated potassium channels (KV), particularly the Shaker channel, providing valuable insights into potassium channel structure and function[227]. κO-PVIIA shares structural similarities with other KV inhibitors, such as charybdotoxin and antitoxins from scorpion venoms. Comparative analysis of its surface charge with charybdotoxin suggests that Lys19 plays a critical role in blocking the channel pore[170].
Table 7. Representative Sequences of κ-Conotoxins[27]
Name |
Sequence |
Target(s) (IC50) |
Ref. |
Piscivorous |
|
|
|
C. purpurascens |
|||
O-PVIIA |
|
meKV Shaker (1616nM) |
[37] |
O-PIVE |
DCCGVKLEMCHPCLCDNSCKNYGK* |
Unknown (No IC50 value) |
[228] |
O-PIVF |
DCCGVKLEMCHPCLCDNSCKKSGK* |
Unknown (No IC50 value) |
[228] |
C. magnus |
|
|
|
A-MIVA |
AOγLVVτAτTNCCGYNOMTICOOCMCTYSCOOKRKO* |
Unknown (No IC50 value) |
[228,229] |
C. radiatus |
|
|
|
M-RIIIJ |
LOOCCTOOKKHCOAOACKYKOCCKS |
hKV1.2 (320nM)> hKV1.3 (1.96μM)> hKV1.1(2.8μM)> hKV1.6 (4.4μM)> hKV1.7 (7.41μM) |
[230] |
M-RIIIK |
LOSCCSLNLRLCOVOACKRNOCCT* |
myKV1.2 (73nM) |
[231] |
Vermivorous |
|
|
|
C. betulinus |
|
||
I2-BtX |
CRAγGTYCγNDSQCCLNγCCWGGCGHOCRHP* |
^BK channels (0.7nM) |
[232] |
C. planorbis |
|
|
|
α/κJ-PIXIVA |
FPRPRICNLACRAGIGHKYPFCHCR* |
moα1β1δε (540nM)>xKV1.6 (1.59μM)> rα3β4 (8.7μM) |
[233] |
C. spurius |
|
|
|
O-SrVIA |
GCGVDGQFCGLPGLGLVCCRGACFLVCIYIP |
xKV10.1 (1.88μM)>xKV11.1 (2.44μM)>xKV1.6 (3.6μM) |
[234] |
C. virgo |
|
|
|
I2-ViTx |
SRCFPPGIYCTPYLPCCWGICCGTCRNVCHLRI |
xKV1.1 (1.59μM)> xKV1.3 (2.09μM) |
[235] |
Notes: PTMs marked in blue bold: Z, pyroglutamate; O, 4-hydroxyproline; *, C-terminal carboxamide; τ, glycosylated threonine; γ, gamma carboxylic glutamic acid; Species: r, rat; h, human; me, common fruit fly; x, X. laevis, m, mouse, my; Cys residues involved in disulfide bonding are shown in red bold. Note: IC50 values were looked for; when not present, other information was used. Tests where no IC50: *, EC50.
KV constitute a highly diverse and abundant ion channel family, comprising approximately 42 different genes organised into eleven subfamilies (KV1 to KV12)[236]. Potassium Shaker channels (KV1 genes) are tetramers, forming functional channels with various subunit combinations, such as homotetramers (e.g. all KV1.2) or heterotetramers (e.g. one KV1.1 and three KV1.2)[236]. Potassium channels are the most diverse group of ion channels, with around 90 different types[237]. They are essential for regulating resting membrane potential, shaping, and timing action potentials, modulating neuronal excitability, and controlling neurotransmitter release. These channels are classified based on the number and arrangement of their alpha-subunits and their activation mechanisms into four main categories: (1) Calcium-gated channels (KCa) featuring six or seven transmembrane segments and a single pore, (2) Voltage-gated channels (KV) with six transmembrane segments and one pore, (3) Two-pore channels (K2P) consisting of four transmembrane segments and double pores, and (4) Inward rectifier channels (Kir) which have two transmembrane segments and one pore[237-239]. This structural diversity enables potassium channels to perform various critical cellular functions. The κ-conotoxins have evolved into multiple families to modulate these channels, with eight recognised classes of K-channel blockers. Interestingly, some μ-conotoxins have also been found to antagonise voltage-gated potassium channels of the KV1 family selectively[3].
3.6.1 κM-RIIIK
κM-RIIK, sourced from C. radiatus venom, belongs to the M-type gene superfamily and exhibits potent and selective inhibitory activity against specific potassium channels[240]. TEVC studies in X. laevis oocytes revealed that κM-RIIIK irreversibly blocks Shaker potassium channels with high affinity (IC50 of 1.21μM). Site-directed mutagenesis studies identified Lys427 as a critical residue within the Shaker channel for κM-RIIIK binding, as mutations to Asp or Asn at this site significantly reduced κM-RIIIK’s potency by over 100-fold[240]. κM-RIIIK demonstrates remarkable selectivity for specific potassium channel subtypes. It does not inhibit other potassium channels, including KV1.1, KV1.3, KV1.4, KV2.2, and KV3.4 at 10μM. Furthermore, κM-RIIIK potently blocks the Tsha1 channel in rainbow trout with an IC50 of 73nM, involving amino acids Leu1, Arg10, Lys18, and Arg19, suggesting a distinct positively charged pharmacophore[241]. In human and rat KV1.2 channels, κM-RIIIK exhibits potent inhibitory activity with ED50 values of 352nM and 335nM, respectively. Importantly, κM-RIIIK shows no inhibition on rat KV1.1., KV1.5, KV1.6 or human KV1.2-KV1.6, KCNQ2/KCNQ3, and BK (KCa) channels at concentrations up to 5-10μM[242]. These findings highlight κM-RIIIK’s unique selectivity for specific potassium channel subtypes, making it a valuable tool for understanding potassium channel function and a promising lead compound for the development of novel potassium channel blockers with therapeutic potential[237].
3.6.2 κM-RIIIJ
κM-RIIIJ is isolated from the same venom as C. radiatus and has been extensively studied recently for its effects on potassium channels. Two-electrode voltage clamp (TEVC) experiments revealed that κM-RIIIJ blocks the rat KV1.1 subtype and human KV1.2, KV1.3, KV1.5, and KV1.6 subtypes with IC50 values of 4μM, 33nM, 10μM, 70μM, and 8μM, respectively[243]. Later, it was identified that the Lys9 residue was crucial for its high affinity to KV1.2, noting that even at concentrations above 70μM, RIIIJ showed no significant inhibition of human KV1.4, KCNQ2/KCNQ3, and KCa channels[243]. In 2019, Cordeiro et al.[244] demonstrated a 100-fold increase in RIIIJ affinity for heteromeric human KV channels composed of three KV1.2 subunits and one KV1.1 or KV1.6 subunit (3:1) compared to homomeric channels, using whole cell patch clamp techniques in HEK293 cells. Due to its ability to selectively bind heteromeric KV channels, κM-RIIIJ has become a valuable tool for identifying neuronal subclasses (L1 and L2), which is particularly important in studying neurological diseases such as multiple sclerosis[237,245].
3.7 Consomatins
Consomatins are a novel, diverse and rapidly growing family of conotoxin somatostatin analogs (Table 8). These evolved receptor ligands mimic native hormones in other organisms, present in fish-hunting cone snails such as C. rolani and C. geographus to effectively incapacitate their prey by disrupting glucose homeostasis. Originating from endogenous somatostatin-like signalling genes, these peptides have undergone significant evolutionary modifications, including incorporating D-amino acids and extensive O-glycosylation, enhancing their stability and receptor selectivity. For instance, Consomatin-Ro1 from C. rolani selectivity actives human somatostatin receptors SSt1 and SST4, exhibiting potent and long-lasting antinociceptive effects comparable to morphine[246]. Similarly Consomatin-nG1 from C. geographus features a minimised somatostatin-like core combined with a heavily glycosylated N-terminal, enabling selective somatostatin 2 (SSTR2) activation in fish prey. This dual-action mechanism, where Consomatins inhibit glucagon secretion while venom-derived insulins induce hypoglycaemia, ensuring sustained disruption of the prey’s glucose regulation, facilitating effect predation called the “ambush and assess hunting”, which was believed to be adapted in response to cone snails hunting dangerous species of fish. In addition to the below species examples, there have been an additional 18 Consomatins across 39 cone snail species through transcriptomic analysis with sequences identities to human somatostatin ranging from 14% to 38%, with a conserved cysteine framework and Tryp7-Lys8 motif essential for receptor binding[45,246].
Table 8. Representative Sequences of Consomatins[246]
Name |
Sequence |
Target(s) (EC50 data) |
Ref. |
Piscivorous |
|
|
|
C. geographus |
|||
Consomatin-G1 |
|
hSSTR2 (2.6nM) |
[45,246] |
Consomatin-G2 |
LFCFwKSCT |
Unknown (No EC50 data) |
[246] |
C. rolani |
|
|
|
Consomatin-Ro1 |
γGYKCVwKTCMOA |
hSSTR1 (2.9μM)>hSSTR4 (5.1μM)
|
[246] |
Consomatin-Ro2 |
ADQTCIwKTWCOOS |
Unknown (No EC50 data) |
[246] |
C. neocostatus |
|
|
|
Consomatin-Nc1 |
MγCYwKSCSRO |
Unknown (No EC50 data) |
[246] |
C. graham |
|
|
|
Consomatin-Gh1 |
ICYwKVCOOSO |
Unknown (No EC50 data) |
[246] |
C. magnus |
|
|
|
Consomatin-Ma1 |
STVPVHICYwKVCOOSOW |
Unknown (No EC50 data) |
[246] |
C. mercator |
|
|
|
Consomatin-Mrc3 |
ALFVPSCIwKTCOY* |
Unknown (No EC50 data) |
[246] |
C. raulsilvai |
|
|
|
Consomatin-Rs1 |
IMFVPSCIwKTCOSY |
Unknown (No EC50 data) |
[246] |
Notes: PTMs are shown in blue bold: O, 4-hydroxyproline; γ, γ-carboxyglutamate; *, C-terminal carboxamide; w, D-tryptophan; Cys residues involved in disulfide bonding are shown in red bold; Cys linkages in black. Species: h, human;
3.7.1 Conus geographus Consomatins
C. geographus was uncovered to have a sophisticated dual-component strategy to incapacitate prey. The venom comprises specialized insulin mimetics (con-insulins) and somatostatin receptor 2 (SSTR2) agonists known as Consomatin-nG1. Consomatin-nG1 is a peptide that exists in multiple O-glycosylated proteoforms. Functional assays revealed that Consomatin-nG1 selectively activates the human SSTR2 with an EC50 of 3.63 nM, exhibiting a bias towards GoA and GoB protein subunits. In isolated mouse islets, 1 nM Consomatin-pG1 administration resulted in a 33% decrease in glucagon secretion under low-glucose conditions (1mM). In contrast, in perfused rat pancreas models, doses of 0.1nM, 1nM, and 10nM Consomatin-pG1 suppressed glucagon secretion by 24% within 5min and reduced glucagon levels by up to 45% at higher concentrations. Consomatin-pG1 demonstrated high selectivity by acting only one of the two homologous SSTR2 receptors, specifically Dr-sstr2b, with an EC50 of 25.32, while showing minimal activity at Dr-sstr2a[45].
The structural analysis of nG1 revealed significant similarities to the somatostatin analogue octreotide, with 80-90% sequence similarity and an EC50 of 2.6 nM at SST2, showing high potency. The native peptide, Consomatin-nG1, also possessed a heavily glycosylated N-terminal tail crucial for its selective activation of SSTR2, a feature that distinguishes it from synthetic analogues. Functional experiments with a des-glycosylated analogue of Consomatin-nG1 showed a significant reduction in potency (EC50=296.4nM) compared to the glycosylated venom fraction (EC50=1.82nM). This indicates that glycosylation is essential for the complete biological activity of Consomatins[45].
3.7.2 Conus rolani Consomatin-Ro1
Consomatin-Ro1 was discovered through a bioactivity-guided fraction of C. rolani venom. Structural analysis revealed that Consomatin-Ro1 contains a disulfide bond between Cys 5 and Cys 10, an N-terminal γ-carboxylated Glu, and hydroxylated Pro at position 12. Notably, the peptide incorporates a D-Trp7 residue, enhancing its stability and mimicking key features of therapeutic somatostatin analogues like octreotide. Functional assays demonstrated that Consomatin-Ro1 selectively activates human somatostatin receptors SST1 and SST4 with EC50 values of 2.9μM and 5.1μM, respectively, while exhibiting negligible activity on other receptor subtypes. Consomatin-Ro1 exhibited remarkable plasma stability, maintaining its integrity for over 158 hours in human plasma compared to 5.5h for native somatostatin-14. In vivo studies in mice revealed that intraperitoneal administration of 2.5mg/kg Consomatin-Ro1 produced antinociceptive effects comparable to 3mg/kg morphine, lasting up to 3h, without significant central nervous system activity when administered intrathecally[246].
4 UNLOCKING THE POTENTIAL OF CONOTOXIN DRUG DEVELOPMENT
4.1 ω-Conotoxin MVIIA (Ziconotide, Prialt®)
The most famous conotoxin discovered is ω-conotoxin MVIIA, also called Ziconotide (Figure 16A). Initially extracted in 1982 from the venom of C. magus, MVIIA was developed into a pharmaceutical agent by Neurex Corp (later Elan Pharmaceuticals) for treating severe pain[247]. The FDA approved Ziconotide in 2004, and it was commercialised under the brand name Prialt®, positioning it as the Principal Alternative to Morphine.
|
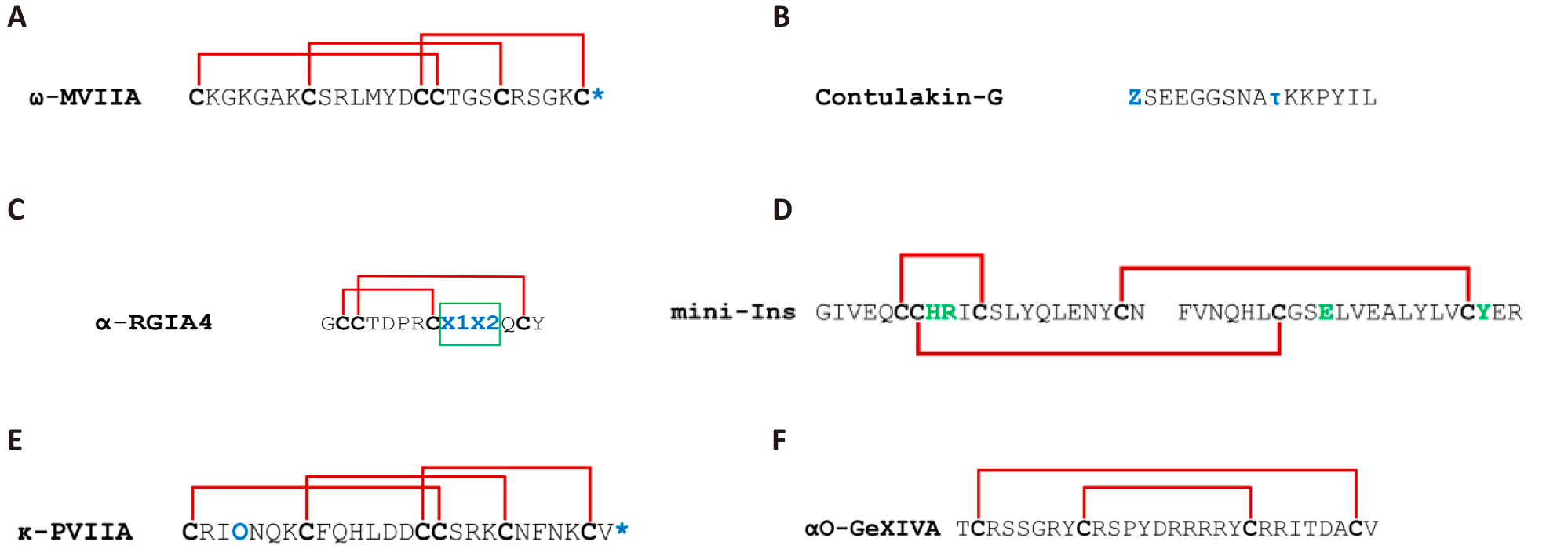
Figure 16. Amino Acid Sequences. A: ω-Conotoxin MVIIA, B: Contulakin-G, C: α-RGIA4, D: mini-Ins, E: κO-PVIIA, and F: αO-GeXIVA. (In bold blue: * C-terminus amidation, Z Pyroglutamate, O 4-hydroxyproline, X1 Citrulline, X2 monoiodotyrosine; In green: modified residues.)
The discovery of ω-MVIIA stemmed from research into the paralytic properties of venom from fish-hunting cone snails. ω-MVIIA has been shown to interfere with neuromuscular transmission in fish by inhibiting a specific type of VGCC at the presynaptic terminus. During the early 1980s, the molecular specifics of calcium channels were not yet fully defined, and the diversity of VGCCs throughout the vertebrate neural systems was unclear. The isolation of ω-MVIIA and ω-GVIA derived from C. geographus provided pharmacological tools for differentiating between VGCCs. The N-type calcium channel, CaV2.2, is the subtype of VGCC that this peptide mainly targets[16].
Neurex Corp's research, which included studies using a radiolabelled analogue of ω-MVIIA, revealed its specific binding to the dorsal root ganglia in the spinal cord layers, critical in pain perception[16,248]. This crucial finding laid the groundwork for developing ω-MVIIA as an analgesic. Although it is a high-affinity, selective ligand, unlike ω-GVIA, ω-MVIIA’s effects are reversible[16]. Consequently, MVIIA is recognised for its analgesic application in treating severe chronic pain, especially with cancer and AIDS, mainly when other treatments are ineffective[15]. In human models, ω-MVIIA has an IC50 value of approximately 6.8nM[36].
Prialt®, the commercial drug, is a synthetic replica of ω-MVIIA. This is a fascinating aspect of Prialt®; no analogue has significantly improved upon the pharmacokinetic properties of the native peptide, resulting in the therapeutic drug being chemically identical to the naturally occurring 25 amino acid product[248]. As a non-opioid agent, Prialt® does not cause addiction or respiratory depression[174]. However, it can lead to serious side effects at high doses, including ataxia and auditory hallucinations, which are usually reversible with dosage reduction[15]. More severe side effects at high doses include debilitating ataxia and psychosis.
Prialt®'s mechanism of action involves specifically targeting CaV2.2 channels in the central nervous system. This necessitates intrathecal administration via an implanted pump into the cerebrospinal fluid. Recently, the high-resolution cry-electron microscopy (cryo-EM) structures of the human CaV2.2. channel, alone and complexed with Prialt®, to reveal how Prialt® selectively blocks CaV2.2 channels. The CaV2.2 complex comprises the central α1 subunit and auxiliary α2δ-1 and β3 subunits. It was shown that Ziconotide nests within the electronegative entrance to the pore and directly engages helices (P1 and P2) and extracellular loops from repeats II, III, and IV of α1, thus sealing off ion access. Notably, the extracellular loop III and α2δ-1 tilt upward to accommodate Ziconotide, which may help explain why α2δ-1 slightly reduces ziconotide’s binding affinity. While three of the voltage sensing domains (VSDs) adopt an ‘up’ (depolarised) conformation similar to those observed in other calcium channel structures, VSDII exhibits a distinct ‘down’ (hyperpolarized-like) state. This was attributed to the two unique cytosolic helices (CH1II and CH2II) and an extended segment of S6 in repeat II that appears to lock VSDII in a downward position. A bound phosphatidylinositol 4,5-bisphosphate (PIP2) molecular lodges within the intracellular side of VSDII, further stabilising its downward shift by preventing the upward motion of S4-5II[249].
Prialt® achieves potent, subtype-specific inhibition of CaV2.2 channels by sterically blocking the pore's outer mouth and neutralizing nearby specific residues. In addition, the VSDII ‘down’ conformation, held by unique CaV2-specific cytosolic segments and PIP2, sheds light on an alternate gating arrangement not previously resolved in eukaryotic voltage-gated Ca2+ or Na+ channels. This structural information opens up the possibility of designing improved therapeutic blockers that might treat chronic or intractable diseases, showcasing how conotoxins can also be crucial pharmaceutical blueprints[249].
This invasive and costly method has limited its broader use, often relegating Prialt® to a last-resort option in clinical settings. However, the scarcity of effective non-opioid treatments has recently led to guidelines promoting Prialt® as a first-line treatment for a range of pain problems, such as nociceptive and neuropathic pain, especially in cancer patients[250]. Also, Prialt® already achieves approximately USD 27 million a year in sales, which is surprising considering that it costs 2.5 times more to manufacture than morphine, with, as a result, it being approximately 2.5 times more expensive in the UK for the National Health Service (NHS) to go through a five-year treatment with Prialt® combination therapy compared to a morphine monotherapy (≈GBP 2.5 million vs GBP 1 million)[251,252]. This remains a substantial limitation for the widespread adoption of Prialt® therapy. Nonetheless, ω-MVIIA being developed into a therapeutic agent exhibits the potential of conotoxins for direct drug development. This was recognised in 2022 with the award of the Golden Goose Award to the discoverers of Prialt®, Craig T. Clark, Lourdes J. Cruz, J. Michael McIntosh, and Baldomero Olivera (See Figure 17, for its 3-D structure)[253].
|

Figure 17. 3-D structure of ω-MVIIA. Reproduced from Ref.[24] with permission from Elsevier.
4.2 Contulakin-G (CGX-1160)
Contulakin-G (CGX) is a unique peptide from the venom of the fish-hunting cone C. geographus, and plays a key role in its “net strategy” for prey capture (Figure 16B). This species, belong to the Gastridium clade, employs CGX to sedate and weaken its prey by releasing it from its rostrum[16]. Notably, mice injected with it intracerebroventricularly showed reduced mobility and responsiveness, earning it its name from the Filipino word “tulakin,” meaning “to be pushed”[16]. CGX is believed to be part of the “nirvana cabal,” a group of toxins employed by net-hunting cone snails to immobilize their prey. Due to their ability to suppress neuronal activity, these toxins, including CGX, hold promise as potential drug leads for conditions such as epilepsy and intractable pain[16].
Unlike typical conotoxins, CGX lacks cysteine residues and disulfide bridges. Discovered for its sedative effects in mice, It is a 16 amino acid peptide with a unique glycosylation at Thr-10 (Figure 16B). Surprisingly, despite lower receptor affinity when initially discovered, glycosylated CGX demonstrated ~10-fold higher potency in vivo that its non-glycosylated counterpart[46]. This result shows the significance of glycosylation in enhancing conotoxin efficacy, potentially through increased stability. the true power of nature’s ingenuity throughout evolution in terms of cone snail venom potency, suggesting that the glycosylation enhanced in vivo efficacy, possibly via increased stability or transport, marking CGX as the first invertebrate neurotensin-like peptide[46].
CGX binds to both mammalian neurotensin receptors isoforms (NTSR1) and (NTSR2)[46]. The use of CGX mirrors its natural role, suggesting it might dampen excessive neuronal activity in fish, like its potential to quiet overactive pain pathways in humans[46]. This contrasts with Ziconotide, as CGX’s natural function is directly linked to its therapeutic value as a non-opioid pain medication. The structure of CGX was deciphered using Edman sequencing, revealing a unique glycosylation at threonine 10[46]. It shares similarities with NT, a neuropeptide involved in dopamine signalling and pain modulation (Figure 18). CGX is a more potent analgesic with lower behavioural toxicity despite lower binding affinity to neurotensin receptors[46]. Its effectiveness and lower agonist activity are attributed to its glycosylation and specific amino acid composition[254].
CGX is a weaker agonist than neurotensin, as reflected by an EC50 of 32nM (vs. 0.8nM for NT)[254]. Because G-protein coupled receptor (GPCR) agonists that bind more weakly are sometimes associated with diminished receptor desensitisation, they evaluated the capacity of CGX and NT to trigger receptor downregulation. In a ‘desensitisation’ assay, pre-exposing cells to different peptide concentrations, then washing and re-challenging with the same peptide, it was discovered that CGX induced 120-fold less receptor desensitisation than neurotensin. Receptor internalization indicated CGX preserved higher cell-surface receptor levels than equimolar NT. These results highlighted how the glycan may reduce binding affinity and minimize desensitization, potentially prolonging analgesic effects[254].
Structure-activity relationship (SAR) data have further explained how O-glycosylation and charged residues shape receptor engagement and downregulation. Notably, removing CGX’s glycan (yielding [T10]Contulakin-G) greatly enhanced its affinity for NTSR1 and altered its desensitization profile. Molecular dynamics simulations (Figure 18) supported the notion that the bulky disaccharide near Thr10 weakens electrostatic interactions with the receptor’s extracellular loops, thus lowering potency. Interestingly, a novel ‘memantine-Contulakin-G’ analog, featuring memantine attached to Glu10, retained Contulakin-G-like weaker agonist activity, showed even less receptor desensitization, and demonstrated analgesic activity in mice after systemic (intraperitoneal) administration[254].
|

Figure 18. CGX-NTSR1 Binding, Glycosylation, and Electrostatics. A: Molecular dynamics simulation showing CGX interacting with the NTSR1 receptor, with its glycosylated (red) and non-glycosylated (yellow) forms depicted using van der Waals. The NTSR1 receptor is in grey. The critical glycosylated residue (Thr10) is highlighted in green. B: The NTSR1 binding site displays the electrostatic potential of the receptor (positively charged in blue, negatively charged in red). Reproduced from Ref.[254] with permission from Frontiers Media S.A.
Licensed to Cognetix Inc. in 2000, the FDA designated CGX as an orphan drug for chronic intractable pain treatment. Early clinical trials and toxicity studies showed promise. Then, CGX entered a Phase Ib clinical trial, which was conducted. CGX entered a Phase Ib clinical study in 2004 for the treatment of chronic intractable spinal cord injury pain at Brigham and Women’s Hospital, Harvard. In a 6-patient study, CGX-1160 showed a significant pain reduction and was generally well tolerated with a broad therapeutic index when administered intrathecally at doses up to 1,000μg/h. Diarrhoea emerged as the dose-limiting toxicity when two subjects receiving 900μg/h developed the adverse effect, establishing this rate as the maximum tolerated dose (MTD). No other significant change in vital signs or adverse events was observed. Cerebrospinal fluid (CSF) sampling revealed a bi-exponential disposition of CGX, featuring a rapid distribution phase and a slower terminal elimination. A pharmacokinetic model incorporating an effect-site compartment best fits the analgesic response, implying a delay between CSF concentrations and peak analgesia. At or below the MTD, CGX produced up to a 63% reduction in spontaneous pain intensity relative to the baseline[255]. These promising results lead to further preclinical studies.
Allen et al.[256] examined in rats and dogs whether administering CGX administered intrathecally or epidurally would yield the best analgesic results. In rats, intrathecal doses (0.03-3nmol) significantly reduced formalin-evoked paw flinching (ED50=0.07nmol), whereas epidural doses (10-89nmol) were less potent (ED50=45nmol). In dogs, intrathecal CGX (50-500nmol) increased thermal skin-twitch latencies in a dose-dependent manner comparable to morphine yet yielded no notable changes in motor function, cardiovascular parameters, or body temperature. By contrast, an epidural dose of 1,000nmol in dogs failed to alter nociceptive thresholds. It also produced no observable motor or autonomic effects at effective doses[256].
Kern et al.[257] followed this up by analysing the intrathecal pharmacokinetics of CGX in beagle dogs under two protocols: a single bolus dose (16.7-1,000nmol) and a 72h infusion (10-160μg/h). Cerebrospinal fluid (CSF) sampling revealed a bi-exponential disposition in both experiments, characterized by a rapid initial phase (on the order of tens of minutes) followed by a slower elimination phase (1-2h). The bolus data indicated a mild nonlinearity at the highest dose (1,000nmol), possibly due to hyperbaric injection or a saturable process at significant concentrations. Infusion experiments, however, yielded lower steady-state concentrations and no apparent nonlinearity[257].
However, the FDA placed the study on partial clinical hold because the preclinical toxicity studies in dogs did not adequately determine the maximum tolerated dose. In 2005, Cognetix shut down when funding was discontinued, and the clinical development of CGX came to a halt despite its very promising therapeutic properties[16].
Recent evidence in rodent models by Martin et al.[258] has advanced our understanding of the antinociceptive properties of CGX, particularly in the context of acute and chronic pain conditions. CGX produced potent antinociceptive effects in rodent models of acute (post-surgical) and chronic (neuropathic) pain, examining its molecular targets and mechanism of action. It was found that NTSR2 activation in presynaptic dorsal root ganglia (DRG) neurons and subsequent blockade of CaV2.3 VGCCs drive CGX’s analgesic effect. CGX was first tested (0.02-0.5nmol, intrathecally) in paw incision and spinal nerve ligation (SNL) rat models, as well as in partial sciatic nerve ligation (pSNL) mice, to replicate both acute and chronic pain states. Dose-dependent reversal of thermal and mechanical hypersensitivity was evident for up to four hours, and efficacy extended to both male and female animals. They employed pharmacological antagonists (SR48692 for NTSR1 and levocabastine for NTSR2) to pinpoint receptor involvement, revealing that NTSR2 blockade negates CGX-induced analgesia, whereas NTSR1 antagonism had no effect. Furthermore, CRISPR/Cas9-mediated known of NTSR2 in mice also abolished CGX’s antinociceptive properties while leaving morphine analgesia intact, confirming that CGX acts predominantly via NTSR2[258].
Subsequent in vitro assays explored CGX’s effect on calcium currents in DRG neurons using whole-cell patch clamps and targeted pharmacological blockers. CGX chiefly reduces CaV2.3 currents (~63% inhibition) with a more negligible effect on CaV2.2., indicating a preferential disruption of CaV2.3 channels. CRISPR knockdown of cacna1e (which encodes CaV2.3) in pSNL mice eliminates CGX-induced analgesia but not morphine efficacy. Additional experiments involving spontaneous EPSC recordings in dorsal horn slices, CGRP release assays, and synaptic fractionation point to a presynaptic mechanism: CaV2.3 is enriched in the presynaptic compartment, whereas NTSR2 localised to both pre-and postsynaptic sites. These results show that CGX triggers an NTSR2-CaV2.3 response, dampening neurotransmitter release from presynaptic terminals in the spinal dorsal horn[258].
This was then followed up by Martin et al.[258] who have extended this approach to cancer-induced bone pain (CIBP), which represents a major clinical challenge often resistant to current analgesics. It is important to note that many patients living with cancer experience severe pain generally poorly managed by conventional treatments. In mice with CIBP, intrathecal CGX does not attenuate spontaneous pain behaviours and evokes mechanical hypersensitivity, independent of sex. CRISPR-mediated editing of NTSR2 or CaV2.3 blocks CGX antinociception. Still, it does not interfere with morphine’s effects. Chronic infusion studies revealed that CGX, unlike morphine, maintains antinociception in CIBP animals with reduced tolerance and without disrupting motor function. Molecular analyses by RNAScope and immunoblotting confirm robust NTSR2 expression in dorsal and ventral horns. However, CaV2.3 appears only minimally in the ventral, potentially explaining why CGX spares motor pathways and remains highly sensory-selective. Taken closely, this refines the NTSR2-CaV2.3 mediated mechanism of CGX action, as mentioned, with CGX already having shown promise in human patients with intractable spinal cord injury-associated pain. It is interesting to see its potential as nonopioid-incident spinal analgesia in both noncancer and cancer pain contexts and highlights its continued promise as a therapeutic candidate for intractable pain states[259]. Taken together, these findings reveal a potential nonopioid pathway for the development of new analgesic agents aimed at treating refractory pain conditions, offering a promising avenue for future interventions[258,259].
4.3 α-RgIA4 (KCP-400) & α-RgIA Analogs
α9α10 nAChRs have a significant role in neuropathic pain. This was discovered with α-conotoxin Vc1.1, an antagonist of these receptors, providing analgesia in various animal pain models, challenging the prior belief that only activation of nAChRs could induce analgesia[260]. This led to the revelation that inhibiting the α9α10 nAChR subtype could be a novel approach to pain treatment. However, subsequent research found that α-Vc1.1 had reduced potency on human α9α10 nAChRs compared to rodent receptors, which would explain poor human clinical trial results[121]. Nonetheless, α-Vc1.1 research paved the way for identifying further α9α10 nAChR antagonists. The crucial finding discovered using cDNA sequencing was α-RgIA from the venom of C. regius[122].
α-RgIA and its synthetic analogs demonstrate significant analgesic and disease-modifying effects in neuropathic pain models. A new analog of α-RgIA, α-RgIA4 (X1, citrulline; X2, monoiodotyrosine) (Figure 16C), showed a high affinity for rodent and human α9α10 nAChRs and effectively prevented chemotherapy-induced neuropathic pain, suggesting its potential for treating cancer-related neuropathies[261]. The lasting analgesic effects of α-RgIA4 also indicated its role in altering the disease state.
More specifically, this is because α-RgIA4 is incredibly specific when inhibiting human α9α10 nAChRs, having an IC50 value of 1.2nM versus 510nM in its native peptide α-RgIA[119,262]. Interestingly, this likely played a role in how, after administration of α-RgIA4 in mice, following treatment with the chemotherapy agent, oxaliplatin, mitigation of cold allodynia induced by the oxaliplatin in mice sustained for a considerable duration up to 21d after administration. This indicates a long-lasting protective effect against the related nerve damage and associated pain with this first-line treatment for colorectal cancer[262].
This is becoming increasingly important as lifespan increases and is associated with the current rise in cancer incidence. Because of these analgesic properties, α-RgIA4 is currently under preclinical development by Kineta Inc., licensed by the University of Utah[16]. α-RgIA4 has also been invested to alleviate paclitaxel-induced neuropathic pain. Rats received paclitaxel injections (2m/kg, i.p.) on days 0, 2, 4, and 6 (total 8mg/kg) and were given daily subcutaneous (s.c.) doses of α-RgIA4 at 16 or 80μg/kg five days per week (days 0-18). Mechanical allodynia was assessed via von Frey filaments. Pain thresholds in the paclitaxel + saline group dropped to a mean of 6.2g (vs 14.6g in controls) by day 16, indicating severe hypersensitivity. Co-administration of α-RgIA4 at 80μg/kg accelerated recovery: by day 31, thresholds increased significantly, and by day 37, mechanical hypersensitivity had markedly improved, notably 12d after RgIA4 cases - suggesting a disease-modifying effect. RgIA4 did not prevent cold allodynia, and paclitaxel did not induce heat hypersensitivity under these conditions. Body weights remained unaffected. In contrast, C57BL/6J mice given the same paclitaxel protocol did not develop robust allodynia or changes in sensory nerve action potentials[263].
This was further investigated by preventing acute oxaliplatin-induced cold allodynia and examining the roles of the α9 and CD3+ T-cells. Adult CBA/CaJ mice received a single intraperitoneal injection of oxaliplatin (10mg/kg), which robustly induced cold hypersensitivity. Daily subcutaneous doses of α-RgIA4 (40μg/kg) significantly prevented this cold allodynia in wildtype mice but did not affect mice lacking the α9-encoding gene (chrna9). Thus, α-RgIA4’s efficacy depends on α9-containing nAChRs. Depleting CD3+ T-cells via anti-CD3ε IgG1 also eliminated the protective effect of α-RgIA4, indicating that CD3+ cells are chosen for its analgesic. Meanwhile, flow cytometric analysis showed that oxaliplatin exposure increased circulating T-cells (CD3+, CD3+CD4+) percentages, α-RgIA4 neither prevented nor significantly altered these elevations. It can be concluded that preventing acute cold allodynia by RgIA4 requires both α9-containing nAChRs and the presence of T-cells. This was found to extend earlier work showing α-RgIA4’s therapeutic potential in neuropathic pain; this provided new evidence that T-lymphocyte and α9-dependent pathways are critical for their protective effects[264].
As outlined, α-RgIA4 holds promise as a non-opioid therapy for neuropathic pain. However, its therapeutic potential is limited by its susceptibility to disulfide scrambling and proteolytic degradation. To address these challenges, it has explored the introduction of a side-chain lactam bridge to stabilize the α-RgIA4’s preferred globular conformation and prevent disulfide rearrangements. Several macrocylic α-RgIA analogs were synthesized using a solid-phase approach involving orthogonal deprotection, lactam bond formation on resin, and controlled cysteine oxidation. Functional assays in X. laevis oocytes expressing human α9α10 nAChRs demonstrated that most macrocyclic analogs retained potent activity, with the lead compound (analog 6) exhibiting an IC50 of 3.4nM, only about twofold less potent than α-RgIA4 itself. Analog 6 exhibited >10μM activity against other human nAChR subtypes except for α7, thus maintaining strong selectivity for α9α10. In a mouse model of oxaliplatin-induced neuropathic pain, daily subcutaneous administration of analog 6 prevented cold allodynia as effectively as α-RgIA4. Further tests of human serum stability revealed that analogue 6 showed significantly reduced disulfide scrambling and higher resistance to proteolysis compared to α-RgIA4. NMR studies demonstrated that the newly introduced macrocycle did not perturb the critical ‘recognition finger’ region essential for receptor binding. A Rosetta-based docking model supported that these macrocyclic variants retain key Arg7 and Asp5 interactions with α9α10 nAChRs[265].
Later, α-RgIA analogues incorporating a methylene thioacetal (-Ch2-) replace one disulfide, stabilizing the peptides while retaining potency for human α9α10 nAChRs. RgIA-5524 has an IC50 of 0.9nM on human α9α10nAChRs, within minimal cross-reactivity against other nicotinic subtypes (>10μM) and only 186nM on α7. Serum stability tests (0.1mg/mL in 90% human serum, 37℃) showed that >70% of RgIA-5524 remained after 24h (vs <25% for α-RgIA4). NMR analysis confirmed that RgIA-5524 keeps the native globular fold (backbone RMSD<0.5Å). Replacing the loop I disulfide abolished activity (IC50=800nM), whereas substituting loop II was well tolerated (IC50=6.1M). Repeated subcutaneous injections (40μg/kg) prevented oxaliplatin-induced cold allodynia; the analgesic effect was lost in α9 knockout mice. This highlights methylene thioacetal as an effective disulfide surrogate[119].
Further engineering of α-RgIA-derived peptides by selectively subsistence-specific cysteines with penicillamine and several non-natural amino acid replacements. The substitution of Cys3 with penicillamine was exceptionally well-tolerated, improving the IC50 from 510nM (native α-RgIa) to 1.3nM for one analogue. Three other cysteine positions could not accommodate penicillamine without drastically losing potency (>1000-fold), and building on these findings and incorporating additional modifications, including β3-homo-tyrosine at residue 13, led to the lead analogue RgIA-5474 with an IC50 of 0.05nM against human α9α10 nAChRs, over 9000-fold better than the native peptide. RgIA-5474 was highly selective and showed no measure inhibition (IC50=>10μM) at other nAChR subtypes, aside from modest activity at α7. This analog also enhanced serum stability, retaining 4% of the active (globular) form in 25% human serum at 24h (versus 64% for RgIA4). When tested in a mouse model of oxaliplatin-induced neuropathic pain, daily subcutaneous dosing of RgIA-5474 (4-40μg/kg) significantly prevented cold allodynia and, notably, pain relief persisted for weeks following treatment cessation. This showed that RgIA-5474 is an ultra-potent, highly stable α9α10 nAChR antagonist and that selectively targeting α9α10 nAChRs with bioengineered conotoxins could provide a promising, non-opioid approach for treating neuropathic pain[266].
4.4 Mini-Ins (Conotoxin Human Insulin Analog)
Mini-Ins is a novel conotoxin-derived human insulin combination developed from full-potency insulin Con-Ins-G1 obtained from C. geographus and licensed to Monolog LLC. It can inspire new fast-acting analogs for human insulin therapy (Figure 16D). Even though the venom-derived peptide Con-Ins-G1, lacking the canonical B22-B30 segment, still binds the human insulin receptor (hIR), they solved multiple crystal structures and performed extensive structure-activity studies[43]. This analog has been designed based on insights from structural biology studies and exhibits unique characteristics that distinguish it from conventional human insulin and its therapeutic analogues[267].
First, Con-Ins-G1 was crystallised with a micro-receptor (μIR) containing the primary insulin-binding site (domains L1, CR, plus an αCT peptide). At 3.25Å resolution, they observed that Con-Ins-G1 engages the hIR surface like human insulin but with fewer atomic contacts. In particular, TyrB15 in Con-Ins-G1 occupies roughly the same hydrophobic pocket that PheB24 occupies in human insulin. Despite lacking the aromatic triplet (PheB24-PheB25-TyrB26), Con-Ins-G1 retains moderate hIR affinity, relying strongly on TyrB15 and, to a lesser extent, TyrB20. Substituting these tyrosines individually with alanine reduced Con-Ins-G1’s potency (for instance, AlaB15 caused a severe drop in Akt activation). Then, it was explored whether human des-octapeptide insulin (DOI), which is monomeric but ~500-fold weaker than native insulin, might be ‘rescued’ by Con-Ins-G1-like substations. Substituting TyrB20 alone into DOI improved its bioactivity fivefold, whereas TyrB15 alone had little effect. Introducing further modifications at A8, A9, and B10 (guided by Con-Ins-G1 differences) ultimately generated a new hIR analogue, mini-Ins, containing four combined: HisA8, ArgA9, GluB10, and TyrB20. Remarkably, mini-ins are fully potent and show receptor-binding affinity on par with human insulin despite lacking the entire B22-B30 region[267].
Using isothermal titration calorimetry, it was shown that mini-Ins binds to the isolated primary site of hIR with ~128-fold weaker affinity compared to human insulin, implying it compensates by having enhanced binding interactions with other receptor domains (e.g., FnIII-1’ loops). Molecular dynamics simulations supported this, indicating stable salt bridges between ArgA9-Glu575 and GluB10-Arg539 in the domain FnIII-1’ region and partial occupation of the hydrophobic pocket by TyrB20. A 1.46 Å crystal structure of min-ins revealed a conventional insulin fold but lacked the extended β-turn. Instead, B20-B22 extends parallel to the B-helix axis[267].
Mini-Ins remains monomeric, verified by analytical ultracentrifugation, and does not elicit antibody response in mice. In rats, mini-Ins lowered blood glucose levels comparably to insulin lispro, and hyper-insulinemic-euglycemic clamp experiments confirmed equivalent potency. The ‘compensation’ strategy evolved by Con-Ins-G1, replacing the canonical PheB24-PheB25-TyrB26 triad with other side-chain interactions, can be translated into a human-based analogue (mini-Ins), offering a promising platform for developing smaller faster-acting insulin therapeutics. It is now proposed that the traditional structure of humans may not be as essential for their biological activity as previously believed. It is still currently in pre-clinical studies[267].
4.5 κO-PVIIA (CGX-1051)
As mentioned, cone snail venoms contain several families of KV channel-blocking conotoxins, which are crucial in the venom’s ability to immobilise prey rapidly. Among these, the conotoxin κO-PVIIA (Figure 16E), derived from C. purpurascens venom, stands out for its potential medical application. This peptide has demonstrated cardioprotective effects in multiple in vivo myocardial ischemia/reperfusion models, offering promising new approaches for treating acute myocardial infarction (AMI). κO-PVIIA has been shown to effectively block Shaker potassium channels, with its interaction with these channels being state-dependent; it binds differently to open channels than to closed ones[268].
In-depth Structure-Activity Relationship (SAR) studies have pinpointed key residues in κO-PVIIA that interact with Shaker channels. The cardioprotective effects of κO-PVIIA were first shown in rabbits[269]. The administration of κO-PVIIA (10 or 100mg/kg) markedly reduced infarction in a rabbit ischemia/reperfusion model when given 5min before reperfusion. However, no protective effects were seen when administered 10 minutes after reperfusion. Isolated heart studies in rabbits indicated that leukocytes do not play a role in this cardioprotection.
Further confirmation of κO-PVIIA’s cardioprotective activity came from studies with rats and dogs[270]. When administered systemically before reperfusion at doses ranging from 30 to 300mg/kg, κO-PVIIA produced a dose-dependent decrease in infarct size in both species. Notably, even at high doses, κO-PVIIA did not significantly affect these animals' blood pressure or heart rate, indicating a substantial safety margin[270]. While the precise mechanism of its cardioprotective action remains fully understood, the finding that κO-PVIIA can prevent the slow inactivation of K+ channels is significant.
Unfortunately, due to the collapse of its patent holder, Cognetix, Inc., after being granted the patent in 2004, κO-PVIIA has never made it past pre-clinical trials (Figure 19 depicts its 3-D structure).
|
Figure 19. 3-D Structure of κO-PVIIA. Reproduced from Ref.[271] with permission from MDPI.
4.6 αO-GeXIVA
4.6.1 Neuropathic Pain
αO-GeXIVA was discovered in 2015, derived from C. generalis, from the South China Sea, and was found to be a potent α9α10 nAChR antagonist (Figure 16F). Three disulfide isomers were synthesized of αO-GeXIVA: the bead isomer (GeXIVA[1,2]), the globular isomer (GeXIVA[1,3]), and the ribbon isomer (GeXIVA[1,4]). Functional assays demonstrated that GeXIVA[1,2] exhibited the highest potency against the α9α10nAChR subtype with an IC50 of 4.6nM, followed by GeXIVA[1,4] with an IC50 of 7nM, and GeXIVA[1,3]) with an IC50 of 22.7nM. When tested across various nAChR subtypes, GeXIVA[1,2] showed significantly reduced potency, with IC50 ratios ranging from 86-fold (mouse muscle nAChR, α1β1δe) to over 1,100-fold (α3β4) compared to α9α10. In vivo evaluations using the rat chronic constriction injury model of neuropathic pain revealed that intramuscular injection of 2nmol GeXIVA[1,2] produced antinociceptive effects equivalent to 500nmol morphine, maintaining efficacy for at least 6 hours without impairing motor function in the accelerating rotarod test. Additionally, NMR analysis confirmed distinct structural features among the isomers, with GeXIVA[1,2] and GeXIVA[1,4] displaying well-resolved spectra indicative of stable conformations, whereas GeXIVA[1,3] exhibited peak broadening suggestive of multiple conformations. This first showed αO-GeXIVA’s potential as a highly selective and potent therapeutic agent targeting α9α10 nAChRs[272].
This later demonstrated that GeXIVA[1,2] effectively mitigates oxaliplatin-induced neuropathic pain in rats. Oxaliplatin-treated rats (n=10) exhibited a significant decrease in mechanical paw withdrawal threshold (PWT) from 20.54±0.80g to 2.71±0.04g and an increase in cold scores from 1.42±0.19 to 5.98±0.15. Single intramuscular (IM) injections of GeXIVA[1,2] at doses of 32nmol/kg, 64 nmol/kg, and 128nmol/kg significantly increased PWT at 2 and 4h post-injection. The area under the curve (AUC) for GeXIVA[1,2] at 128nmol/kg was comparable to 50mg/kg gabapentin, while 64 nmol/kg also showed significant efficacy. Repeated daily IM injections of 128nmol/kg GeXIVA[1,2] over 16d resulted in a cumulative analgesic effect, significantly increasing PWT from day 7 to day 16 and maintaining analgesia up to 10 days post-treatment. The 64nmol/kg dose showed significant pain relief by day 16, whereas 32nmol/kg had minimal effect. A single 32nmol/kg had minimal impact in cold allodynia. In terms of cold allodynia, a single 32nmol/kg dose of GeXIVA[1,2] alleviated symptoms at 6h, and repeated treatments at 32nmol/kg and 128nmol/kg significantly reduced cold scores by day 8. GeXIVA[1,2] did not affect normal pain perception, hind limb grip strength, or body weight in untreated rats after repeated administrations, indicating a reasonably favourable safety profile. This showed how GeXIVA[1,2] offers a potent and selective therapeutic approach for managing chemotherapy-induced neuropathic pain without adverse motor or systemic effects[273].
GeXIVA[1,2] efficacy has been investigated in mitigating chemotherapy-induced peripheral neuropathy (CIPN) in C57BL/6 mice. Neuropathy was induced through intraperitoneal injections of oxaliplatin at 3.5mg/kg/day for weeks (15 injections). Concurrently, mice received subcutaneous injections of GeXIVA[1,2] at doses of 0.0865mg/kg, 0.692mg/kg, and 1.73mg/kg daily, with control groups administered normal saline. Behavioural assessments using the von Frey test revealed that the highest dose of 1.73mg/kg completely prevented the development of mechanical allodynia, maintaining the paw withdrawal threshold (PWT) compared to saline controls. In contrast, the 0.692mg/kg dose significantly alleviated allodynia at week 3, and the 1.73mg/kg dose was effective at both weeks 2 and 3. Additionally, open field test (OFT) and elevated plus maze (ELPM) evaluations indicated that GeXIVA[1,2] at 1.73mg/kg/day for 3 weeks had no adverse effects on locomotor activity or anxiety-like behaviours. RNA sequencing of the L4-L6 spinal cord identified 209 differentially expressed genes (DEGs) in the CIPN model, with 106 up-regulated and 102 down-regulated compared to controls. Treatment with GeXIVA[1,2] altered 346 DEGs (233 up-regulated and 113 down-regulated) compared to the OXL + NS group, and 53 overlapping DEGs were significantly modulated by GeXIVA[1,2], predominately within immune-related pathways such as cytokine-cytokine receptor interactions. qRT-PCR validation confirmed that genes like Neb were significantly up-regulated in the CIPN group and down-regulated following GeXIVA[1,2] treatment. Furthermore, chronic GeXIVA[1,2] injection in normal mice resulted in 385 DEGs (302 up-regulated and 83 down-regulated) without affecting normal pain thresholds or behaviours[274].
4.6.2 Anti-Cancer Properties
The anti-cancer properties of αO-GeXIVA were first discovered in cervical cancer cell lines SiHa and CaSki, which exhibit significant overexpression of the α9 and α10 nAChR subunits, with SiHa cells showing a 1.80-fold and CaSki cells a 1.50-fold increase in protein levels compared to normal ectocervical cells (Ect1/E6E7). Additionally, α3, β2, β3, and β4 nAChr subunits were upregulated in SiHa cells, with the β4 subunit being 3.12 times higher than in normal cells. Treatment with αO-GeXIVA, a selective α9α10 nAChRs, significantly inhibited the proliferation of both SiHa and CaSki cancer cells in a dose-dependent manner across concentrations ranging from 12.5μM to 200μM, which resulted in statistically significant reductions in cell viability in cancer cells while exhibiting considerably lower inhibition rates on normal Ect1/E6E7 cells[275,276].
αO-GeXIVA has also been found to impede the proliferation of breast cancer MDA-MB-157 cells by targeting α9 nAChRs. The cell counting Kit-8 (CCK-8) assay revealed that GeXIVA inhibited MDA-MB-157 cell viability with an IC50 of approximately 78.31μM after 24h of treatment while exhibiting minimal toxicity towards normal breast epithelial cells (HS578BST). Flow cytometry analysis showed that αO-GeXIVA treatment increased the proportion of apoptotic cells from 0.73% in the control group to up to 27.05% at higher concentrations (11.25-90μM). Additionally, αO-GeXIVA induced S-phase cell cycle arrest, significantly increasing the number of cells in the S phase while decreasing those in the G0/G1 and G2/M phases. The wound healing assay demonstrated that αO-GeXIVA reduced the migration distance of MDA-MB-157 cells in a concentration-dependent manner, with migration distances decreasing from 24.52% in control cells to 17.19% at 90 μM after 24h. Stability tests using RP-UPLC indicated that αO-GeXIVA remained approximately 93% intact in serum-free DMEM after 24h, decreasing to 54% in 10% GBS over the same period. Furthermore, the establishment of an α9-nAChR knockout (KO) MDA-MB-157 cell line via CRISPR/Cas9 revealed that αO-GeXIVA anti-proliferative effects were abrogated in KO cells, with survival rates remaining high (1.2% to 18% inhibition in wild-type versus minimal effect in KO cells at 90 and 180μM)[277].
Guo et al.[278] also investigated the effect of 0.1 nmol per mouse (equivalent to 17μg/kg) of GeXIVA[1,2] daily for 10 consecutive days in a 4T1 mouse allograft model of triple-negative breast cancer (TNBC). This treatment resulted in a significant shrinkage of tumor volumes and a marked reduction in tumor weights compared to the saline control group without causing any notable changes in the mice’s body weights, indicating minimal toxicity. In vitro, GeXIVA[1,2] exhibited potent cytotoxicity against 4T1 cells with IC50 of 62.52μM at 48h and 23.99μM at 72h. Western blot analyses demonstrated that GeXIVA[1,2] treatment upregulated the Bax/Bcl-2 ratio and increased the cleavage of Caspase-3, thereby inducing apoptosis. Additionally, GeXIVA[1,2] significantly downregulated phosphorylated AKT (p-AKT), phosphorylated mTOR (p-mTOR), phosphorylated STAT3 (p-STAT3), and NF-κB expression levels, while phosphor-ERK (p-ERK) and phospho-JNK (p-JNK) remained unchanged. This suppressed TNBC tumor growth in vivo by inducing apoptosis and inhibiting key proliferative signalling pathways at a low, non-toxic dose[278].
5 FUTURE DIRECTIONS AND CONSIDERATIONS
Conotoxins hold immense potential in drug development, but clinical applications face several challenges. Emerging directions in the field show promise in overcoming these obstacles. This section highlights these innovations, potential in silico-based therapeutic breakthroughs, and essential considerations for sustainable conotoxin research and bioprospecting.
5.1 Application of AI-based Tools
5.1.1 Transcriptome Mining: ConusPipe
ConusPipe is one of the first revolutionary machine learning-based toolkits applied for conotoxin research. The most modern method for discovering novel conotoxins involves transcriptome sequencing of Conus venom glands, followed by de novo assembly and homology-based toxin identification[279]. However, homology-based searches are limited to sequences similar to known conotoxins. ConusPipe overcame this by predicting potential conotoxins based on their chemical characteristics, using three machine learning models: (i) Logistic regression, (ii) Semi-supervised learning (label spreading), and (iii) An artificial neural network (perceptron). This was used in conjugation alongside cross-species Blastp searches to maximise discovery potential[280].
ConusPipe uncovers novel conotoxin transcripts for cone snail venom gland transcriptomes, even when they exhibit no detectable similarity to known conotoxins. Existing search strategies relied heavily on homology-based methods (e.g., BLAST or HMMER), which only detect sequences that resemble known toxins. To transcend these limitations, ConusPipe focuses on prominent chemical traits shared across conotoxins: an N-terminal signal sequence, a mid-region propeptide sequence, and a single mature toxin region at the C-terminus. Three supervised or semi-supervised classifiers (logistic regression, label spreading, and a single-layer perceptron) with a cross-specific BLAST approach. Each classifier captures different aspects of conotoxin features, and ConusPipe allows users to merge their outputs (via unions or overlaps)[280].
To train and cross-validate models, 4,950 confirmed conotoxin sequences from ConoServer/UniProt and 52,613 non-conotoxin transcripts. They extracted 15 continuous features, including the SignalP D-score, cysteine percentage, and charge composition for each of the three conotoxin regions. Ten-fold cross-validation demonstrated high predictive performance, with label spreading achieving up to 90.93% sensitivity and 99.07% specificity, and logistic regression and perceptron also showed robust accuracy above 96%. Cross-species BLAST was applied to reduce potential toxins that lacked complete structural motifs or diverged so significantly that they fell below the machine-learning thresholds. Benchmarking on known conotoxin superfamilies showed that the best sensitivity (around 95.7%) was achieved by taking the union of three or four methods. In comparison, the highest specificity (nearly 99.9%) occurred when requiring an overlap of multiple methods[280].
They analysed 12 RNA-seq datasets (757,932 transcripts) representing 10 Conus species to discover new conotoxin candidates. After discarding sequences with existing matches in public databases (Uniprot/ConoServer), ConusPipe predicted 5,479 new toxin-like transcripts, which were further filtered by BLAST searches against NCBI’s non-redundant database to remove any that matched known proteins. Ultimately, 5148 transcripts remained, showing no homology to published toxins but bearing hallmark conotoxin features. Of these, 896 were corroborated by at least three of the four classification methods, pointing to very high-confidence predictions. Single-linkage clustering revealed hundreds of potential novel conotoxin superfamilies, thus significantly broadening the known conotoxin. ConusPipe offers a decisive step toward discovering novel conotoxins, establishing an essential resource for transcriptome-mining pipelines and accelerating the search for new pharmacologically active peptides[280].
5.1.2 Conotoxin Prediction & Generation
With the amount of conotoxin mature peptide sequences able to be sequenced and generated, this has raised some interesting questions on whether using existing conotoxin structural and experimental data, using Artificial Intelligence (AI) and Machine Learning (ML) further, discovering a novel conotoxin or trying to discover a more potent analog, that this could be achieved skipping having to perform structural biology and electrophysiology studies, essentially get straight to the answer.
5.1.2.1 Sequence-Based Prediction of Class and Target
Monroe et al.[281] have pioneered machine learning to predict the target receptors of conotoxins based solely on their amino acid sequences. Conotoxin identification is typically hindered by their complex structures, including multiple disulfide bonds, various PTMs, and inherent inflexibility, which significantly impact their toxicity and receptor binding. To overcome these challenges, a novel ML approach that incorporated 13 unique features across four categories was developed: (i) Primary sequence-based features (P), (ii) PTMs (P2), (iii) Structural properties (SS), and (iv) collisional cross-section (CSS) values from ion mobility-mass spectrometry (Figure 20A). These features provided a more comprehensive understanding of conotoxin physiochemical properties, secondary structures, disulfide bond arrangements, and overall molecular topology. Utilizing a curated dataset: positive (conotoxins), easy-negative (non-toxic peptides), and hard-negative (other toxic peptides), it was demonstrated that integrating these features significantly improved the accuracy of several ML classifiers, including Penalized Logistic Regression, Support Vector Machines, Random Forest, and XGBoost. Notably, combining all feature categories achieved an impressive overall accuracy of approximately ~96% and F1 scores of around ~0.92, surpassing the performance of existing prediction tools like ToxinPred and TOXIFY. This advancement addressed the limitations of previous ML models and effectively handed imbalanced datasets through techniques like Geometric SMOTE. Figure 20B presents a workflow of this process[281].
|
Figure 20. Feature Group Analysis and Conotoxin Predication Workflow. A: The features were categorized into four distinct groups (P, P2, SS, and CCS), and the impact of each group on the accuracy of conotoxin prediction was systematically assessed. B: Workflow for the process. Reproduced from Ref.[281] with permission from MDPI.
Building upon the work of Monroe et al.[281], Truong et al.[282] refined the ML model by incorporating the SMOTE-Tomek hybrid technique to address the challenges of limited and imbalanced datasets. This enhanced model established significant improvements. For conotoxin class prediction (α, μ and ω), the Penalized Logistic Regression model with Principal Component Analysis (PCA) under the SMOTE-Tomek framework, trained on both primary sequence (P) and structural (SS) features, achieved the highest performance with approximately 96% overall accuracy and 93% average accuracy. In predicting the nAChR-binding conditions, the SMOTE-Tomek PCA Support Vector Machine (SVM) model, utilizing P and CCS features, yielded an accuracy of around 91%. These findings, illustrated in Figure 21 A to B, further emphasized the crucial role of structural features in capturing the functional signatures of conotoxins[282].
|
Figure 21. SMOTE-Tomek Data Balancing and ML Pipeline. A: This cartoon illustrates the SMOTE-Tomek technique for handling imbalanced datasets. SMOTE (Synthetic Minority Over-Sampling Technique): New synthetic data points are generated for the minority class by interpolating between existing minority class samples. Tomek Links: Identifies and removes borderline samples (red circles) (instances close to the decision boundary between classes) to improve class separation. This combined approach helps to balance the dataset and enhance the performance of the ML model. B: An overview of the ML pipeline used by Truong et al[282]. It outlines the steps in training and cross-validating the classifier, including data preprocessing, feature selection, model training, and evaluation. Reproduced from Ref.[282] with permission from MDPI.
5.1.2.2 In Silico Conotoxin Discovery
ConoDL is a recently unified end-to-end deep learning framework that rapidly generates and filters conotoxin-like sequences. ConoDL comprises two main modules: ConoGen, a Transformer-based generative model built by fine-tuning a large language model (ProGen), and ConoPred, a prediction model based on a Wasserstein Autoencoder (WAE) architecture. To train ConoGen, the authors curated 2,310 mature conotoxin sequences from the ConoServer database, filtering out non-natural amino acids and focusing on peptides 10-70 amino acids long. ConoGen then leveraged the extensive parameter space of the ProGen model, which was initially pre-trained on 2.81 billion protein sequences spanning around 19,00 protein families. Fine-tuning ProGen on these conotoxin data addressed the challenge of having relatively few known conotoxin sequences (roughly 4,670 in total, reduced to 2,310 after strict filtering) while also enabling ConoGen to capture the complex sequence features that characterized cysteine-rich conotoxins[283].
By sampling from ConoGen’s autoregressive next-residue prediction, 1 million artificial conotoxins sequences were generated in under ten minutes, finally retaining 761,371 unique sequences after duplicate removal. ConoPred was trained with 2,310 positive examples (genuine conotoxins) and 13,941 negative examples (general proteins with no conotoxin annotations) to distinguish possible conotoxins from low-likelihood sequences. ConoPred’s bidirectional GRU encoder and WAE architecture mapped sequences into a 100-dimensional latent space, and the subsequence classifier reported the probability of each sequence being a conotoxin. On both training and test sets, ConoPred achievers near-perfect discrimination (AUC=1, AUPR=0.998). When applied to 439 conotoxin sequences excluded from training, the model scored most above 0.9, whereas 11,531 non-conotoxin UniPro sequences cored predominately below 0.10. The ConoGen-generated sequences mostly scored above 0.80, suggesting that the framework successfully learned conotoxin-like features[283].
From the sequence perspective, the generated peptides strongly resembled natural conotoxins in length distribution (mainly 20-60 residues), amino acid composition, net charge, and overall hydrophobicity. Approximately 35.8% of the artificial sequence exhibited one of 29 known cysteine frameworks, while 64.2% appeared to form novel scaffolds, suggesting that ConoGen can replicate known disulfide bonding patterns and propose new ones. In terms of similarity, alignment scores indicated that ConoGen’s outputs shared moderate levels of overlap within the original training set, showing an average similarity score near 10.86 compared to natural conotoxins and 15.58 among themselves, consistent with a balance between learning conotoxin features and introducing new variations. Most artificial-natural sequence identities clustered around 25-45%, peaking at about 35%, highlighting that the model is not merely memorising known conotoxins but generating novel, conotoxin-like peptides[283].
These artificial sequences can form appropriate disulfide bonds and fold into stable three-dimensional structures. This can be confirmed using ColabFold (AlphaFold2) to predict 34 distinct peptide structures from 30 cysteine scaffold categories and perform 500 ns of all-atom molecular dynamics (MD) simulations on 37 different artificial conotoxins. In 31 MD simulations, Cα RMSD remained below 3 Å throughout the runs, indicating stable conformations consistent with the AlphaFold2-predicted structures. The remaining six trajectories, fluctuating between 4 and 6 Å in RMSD, did so mainly in regions lacking disulfide bonds, suggesting that the cysteine-bridged cores remained stable while other segments allowed more conformational freedom[283].
It appears that ConoDL has serious potential to facilitate large-scale virtual conotoxin discovery and accelerate the development of novel bioactive peptides in related cysteine-rich families. Future interactions will include expanded functionality, such as predicting disulfide bond connectivity and the peptide's potential receptor targets. If this is accomplished, it likely has the potential to revolutionize the field[283].
5.2 Advances in Human Receptor Computational Modelling
Human receptor computational modelling advances have greatly improved our understanding of conotoxin pharmacology. Human tissue models provide insights into conotoxin interactions with human biological systems, leading to safer and more efficient drug development[16]. Consequently, much of the existing research on conotoxins likely must be revisited using human receptor modelling and direct human receptor studies. This is particularly important as findings and case studies suggest that conotoxins toxic to animals, such as rats, may not necessarily exhibit the same toxicity in humans, as illustrated in the Tables in Section 3 and the case study of α-Vc1.1 in Section 4.3[1,196,207]. Despite conotoxin's therapeutic promise, realising their full pharmacological potential requires considerable time and resources through extensive structure-activity relationship (SAR) studies. However, leveraging computational tools to predict mutational effects is anticipated to streamline their design, enabling more efficient development of human subtype-specific ligands[117,284].
5.2.1 Case Study: Engineering α-PeIA for the hα6/α3β4 subtype
As outlined, nAChRs, especially the α6/α3β4 subtype, are key targets for neuropathic pain treatment[285]. However, sensitivity differences between rodents and human nAChRs complicate using rodent models for therapeutic screening. Hone et al.[117] examined the interactions between α-conotoxin PeIA and α6/α3β4 nAChRs to develop a peptide variant with comparable potency for human and rat receptors[117].
Molecular dynamics simulations revealed distinct ligand-binding motifs and key residues contributing to differential sensitivity[117]. Most residue interactions were between species, but non-conserved amino acids in the β4 subunit (positions 36, 119, 163, and 168) were species-specific. These findings emphasised the importance of the second loop of α-PeIA (residues 9-16) in establishing different interactions with the α6/α3β4 nAChRs[117].
To validate these insights, α-PeIA analogues with specific substitutions were synthesised and tested for potency on human and rat α6β4 nAChRs expressed in Xenopus laevis oocytes. Functional assays confirmed that human and rat α6/α3β4 nAChRs exhibit markedly different sensitivities to α-conotoxins, reflecting the species-specific ligand-binding motifs identified in simulations[117].
One key outcome was the development of PeIA-5667 (Figure 22A), a peptide variant with 270-fold higher potency for rat α6/α3β4 nAChRs (IC50=0.48nM) than the native α-PeIA (IC50=130nM), while maintaining similar potency for the human homologue (IC50=0.2nM)[117]. PeIA-5667 showed a subnanomolar affinity for both human and rat α6/α3β4 nAChRs, indicating its potential as a therapeutic ligand for neuropathic pain treatment. This variant was created by incorporating specific substitutions (A7V, S9N, N11R, and L15I) identified as critical for enhancing binding affinity in both human and rat receptors[117].
|

Figure 22. α-PeIA/PeIA-5667 Alignment and Variant Modifications. A: Sequence alignment of PeIA and PeIA-5667. Cysteine residues in bold, linked with red disulfide bridges, green for modified amino acids (A7V, S9N, N11R, and L15I), and bold blue for PTMs). B: Sequence alignment of PeIA and its four variants was developed by Wu et al[284].
Mechanistic insights included the A7V substitution introducing a longer side chain and increasing contacts at the peptide-receptor interface, particularly with the rat receptor[117]. The S9N substitution created new hydrogen bonds that enhanced binding affinity, while the N11R substitution improved binding through charge interactions with the receptor’s binding site negatively charged environment.
The development of PeIA-5667 stresses the utility of combining computational modelling with functional assays to overcome the limitations of traditional homology-based methods[117]. By mapping the chemical space of the α6/α3β4 nAChR ligand-binding site and identifying fundamental interactions, it was demonstrated that ligands with similar potencies for human and rat α6/α3β4 nAChRs can be generated[117]. This approach enhances our understanding of species-specific differences in receptor pharmacology and provides a framework for designing effective therapeutic ligands across species.
5.2.2 Global Applications: Advances in α-Conotoxin Engineering
Wu et al.[284] expanded this work by applying computational methods to five α-conotoxins. They developed molecular models for six nAChR subtypes in complex with five α-conotoxins (PeIA/α3β2, PeIA/α6β2β3, TXID/α3β4, TxID/α6β4, BuIA/α6β2β3, BuIA/α6β4, ImI/α7, and RgIA/α9α10)[284]. These models were refined using energy minimisation (EM) and molecular dynamics (MD) simulations, providing information about the structural stability and interactions of α-conotoxins with their targets[284].
Wu et al.[284] used five mutational energy prediction methods: (i) FoldX, (ii) BeAtMuSiC, (iii) Coarse-Grained Umbrella Sampling (CG_US), (iv) Molecular mechanics Poisson-Boltzmann surface area/Molecular mechanics generalized born surface area (MMPBSA/MMGBSA), and (v) ToxDock, evaluating their accuracy in predicting changes in binding affinity upon mutations[284]. FoldX combined with MD simulations emerged as the most effective, achieving an accuracy of 85% and a Matthews correlation coefficient (MCC) of 0.68[284]. This combination demonstrated superior predictive power, particularly after structural refinement, outperforming other methods like BeAtMuSiC and ToxDock, which showed moderate accuracy, and CG_US and MMPBSA/MMGBSA, which lacked predictive reliability due to their coarse-grained approaches. The only exception for Foldx was the BuIA/α6β4 system with an accuracy of 33.3% and MCC of -0.25[284].
Experimental, computational predictions used synthesised α-conotoxin mutants; α-PeIA was used previously, so this one was selected, and its prediction was comparable to the overall average prediction statistics. Four variants of PeIA (H5E, S9F, P13G, H12V) were synthesised and tested using two-electrode voltage-clamp electrophysiology in Xenopus laevis oocytes in rat α3β2 and α6/α3β2β3 nAChRs (Figure 22B)[284]. Predictions for changes in activity aligned closely with experimental data, even predicting decreased activity at both nAChR subtypes (H5E and H12V), demonstrating how robust this computational framework has developed. For example, the S9F mutant exhibited enhanced activity at α6/α3β2β3 (IC50 2.0 nM) while maintaining comparable activity at α3β2 (IC50 2.4 nM), consistent with computational forecasts[284]. However, some inaccuracies arose, particularly for mutant H12V at α6/α3β2β3, which induced significant backbone conformational changes, which the computational models did not fully capture[284]. These deviations highlight the limitations of FoldX and current prediction software when significant structural alterations occur but highlight its effectiveness for systems with minimal backbone changes[284].
Advances in human receptor computational modelling and α-conotoxin engineering hold implications for further conotoxin in silico drug discovery. Designing conotoxin therapeutics more efficiently by integrating computational tools with experimental approaches could significantly reduce time and costs. This innovative framework provides detailed insights into mutational effects and offers a pathway for creating targeted therapeutic strategies.
5.3 Regulatory Hurdles
Conotoxin research faces substantial regulatory challenges due to biosecurity concerns and their diverse and complex chemical structures and biological activities[1]. Several global regulatory framework bodies maintain lists of regulated substances, including pathogens and toxins, but conotoxins are often broadly listed without specific classification[1]. This complicates their regulation, as seen in the European Union and Australia, which list “conotoxins” without much differentiation, like the United States' approach before a 2012 revision[1,286]. This broad categorisation includes toxic and non-toxic conotoxins and those with unknown biological activity, presenting significant regulatory hurdles.
In response, some countries have refined their definitions[1]. The United States, for example, revised its select agent list in 2012 to include only paralytic α-conotoxins with specific sequence motifs that block muscle-type nicotinic acetylcholine receptors[287]. This precise classification can help mitigate the issues arising from broad categorisations. Australia has refined its definition of conotoxins ‘in product form’, meeting three criteria: (i) Are pharmaceutical formulations designed for human administration to treat medical conditions? (ii) Are pre-packaged for distribution as medical products. (iii) Are authorised by a state authority to be marketed as medical products[286].
The Australia Group, established in 1985, includes 44 members collaborating to harmonise export controls and reduce the proliferation risk of chemical and biological weapons[288]. Conotoxins are listed as biological agents, requiring controlled international trade, except for medical or clinical formulations for human use. Regulations vary across countries, with researchers typically allowed to work with threshold amounts of conotoxins under less stringent requirements, while larger quantities face rigorous controls. Regulatory agencies usually focus on potent paralytic toxins, but the lack of specific definitions often leads to all conotoxins being regulated similarly, regardless of their toxicity[1].
Interestingly, crude cone snail venom, even from highly venomous species, is not regulated; only individual conotoxin components are controlled. Despite crude venom potentially being more toxic due to synergistic effects, regulatory measures reflect concerns about the misuse of conotoxins in bioterrorism. However, no documented misuse exists outside legitimate research and drug development[1].
Concerns include the potential for aerosolising conotoxins and facilitating their spread and inhalation, though formulating peptides for aerosol delivery is technically challenging[1,289]. Another concern is the use of conotoxins as injectable murder weapons, given their potency in small amounts. The difficulty in detecting these small quantities in forensic investigations and the need for antivenom further complicates potential medical responses. Additionally, there is concern about incorporating conotoxins into pathogen genomes to enhance their lethality, as alleged in historical bioweapon programs[1].
The lack of a clear definition for “conotoxin” is problematic. Regulatory authorities should distinguish between different conotoxins to ensure appropriate control measures. Listing even the most potent toxins may have little impact on bioterrorism prevention, as information on toxin sequences and synthesis methods is widely available. Restricting research on these toxins could hinder scientific progress[1].
Broadly, conotoxin regulation must balance biosecurity concerns with the need for scientific research. More precise definitions and targeted regulations can help achieve this balance, enabling researchers to explore the pharmacological potential of conotoxins while mitigating the misuse risks[1].
5.4 Conservation
The promising field of conotoxin research comes with a responsibility to conserve the habitats of cone snails. Protecting the marine ecosystems where these snails thrive is crucial for sustaining conotoxin research, maintaining ecological balance, and harnessing their vast pharmacological potential. Given that many cone snail species are in the developing world (Figure 23), it is essential to emphasise their conservation.
|
Figure 23. The Global Distribution of Cone Snails Is Indicated by Colour-Coded Spots Representing the Number of Species Present in Each Area. Reproduced from Ref.[290] with permission from Elsevier.
5.4.1 Global Cone Snail Population Conservation Assessment
A Conservation Assessment of the status of 632 Conus species, using the International Union for the Conservation of Nature Red List standards, revealed that three-quarters (75.6%) of the species are not currently at risk of extinction due to their wide distribution and perceived abundance. However, 6.5% are threatened with extinction, and 4.1% are near threatened. Data deficiency prevented categorisation of 13.8%, though they possessed concerning characteristics[291].
Notably, 42.9% of the 98 species were classified as threatened or near threatened in regions with hotspots of endemism, such as the Eastern Atlantic. All 14 species categorised as Critically Endangered and Endangered are endemic to either Cape Verde or Senegal, with the three Critically Endangered species restricted to single islands in Cape Verde. The threats to these species are primarily driven by habitat loss and anthropogenic disturbances, including urban pollution, tourism, and coastal development. The extinction risk faced by cone snails is comparable to that of many well-assessed terrestrial taxa, challenging the belief that marine species are less at risk[291].
Threats to Conus species at risk of extinction vary and depend on the proximity and nature of human habitation and development adjacent to their coastal habitats. Wide-ranging species can maintain viability through resilience from multiple sub-populations, while range-restricted species are more vulnerable. Of the 41 Conus species globally assessed as threatened with extinction, 32 (78%) occur within an area of occupancy (AOO) of 250km² and a minimum depth of 5 meters or less. In the Eastern Atlantic, this figure rises to 100%[291].
Threats to these species can be classified into four main groups: (i) pollution from petrochemical spills or urban and industrial effluents, (ii) habitat disturbance from coastal development, (iii) shell gathering, and (iv) environmental changes such as elevated sea-surface temperatures. Often, these threats are compounded by factors such as tourism infrastructure, which can increase shell gathering, and the proximity of unregulated purlieus, which elevate the risk of effluent discharge into the marine environment. Habitat destruction from activities like sand removal, beach nourishment, and recreational sea use also poses significant threats to local mollusc populations[291].
Cape Verde is undergoing a significant change from a mainly services and fisheries-based economy to one focused on beach tourism[291]. This shift brings numerous threats from road and resort construction, unlawful sand removal for cement, and casual shell gathering by tourists. The three critically endangered species in Cape Verde are already experiencing population decline due to habitat loss and development. For example, C. salreiensis, restricted to a single bay, has seen observable declines since constructing a harbour[291].
In Senegal, species restricted to the highly polluted coastal waters around the Dakar peninsula show a marked decline in abundance and shell size. This decline is attributed to the burgeoning population and inadequate waste-processing infrastructure. In Angola, Conus species face similar threats[291].
In the Western Atlantic, coastal development and tourism have led to significant habitat loss for species like C. anabathrum and C. stearnsii along Florida's coast. Tourism threatens species like C. hennequini in Martinique and C. hieroglyphus in Aruba. Shell collecting in the Bahamas threatens C. richardbinghami, while coastal development in Bahia, Brazil, threatens C. henckesi. The Venezuelan government’s plans for development on the islands of Los Roques could place the shallow water species C. duffyi at risk[291]. In Southern Natal and the Mascarene Islands, species have declined due to over-collecting and intensive trawling, significantly reducing their populations and genetic diversity[291].
The assessment also highlighted the issue of data deficiency, with 87 species (13.8%) categorised as Data Deficient. Many of these species are considered scarce in the wild, though the causes and extent of their threats are poorly understood. Data Deficient species often have restricted ranges and are found at greater depths, making them less studied[291].
This global assessment of Conus species highlights the need for targeted conservation strategies. This includes improved data collection, sustainable shell collecting practices, and strengthened Marine Protected Areas (MPAs) enforcement. With its high percentage of endangered species, Cape Verde represents a critical area for conservation efforts, necessitating stringent export controls and comprehensive environmental impact assessments for new developments[291].
5.4.2 Safeguarding Cone Snail Biomedical Potential
Traditionally, scientific studies on cone snails have concentrated on widely distributed species, resulting in limited impact from research-related collection. Despite concerns about the potential overharvesting of cone snails for research, advances in analytical methods, such as increased sensitivity and high-throughput next-generation screening (NGS) technologies of the venom duct transcriptome, now allow researchers to derive extensive data from minimal samples. Most laboratories today collect only a few specimens per species annually; in some cases, a single snail can provide sufficient material (Figure 24)[292].
|
Figure 24. Current Approaches for Maximising Scientific Insights from a Single Specimen. This study demonstrates how a single cone snail specimen can yield extensive data across multiple domains, including genomics, transcriptomics, and proteomics. Reproduced from Ref.[292] with permission from Nova Science Publishers.
Venom collection relied on dissecting and extracting tissue from the venom glands of frozen cone snails[30,293-295]. While this method yielded results, it involved sacrificing large numbers of snails. An alternative approach, venom ‘milking’, extracts venom from live snails without harming them. Milked venom offers several advantages over venom obtained through dissection. It is cleaner, contains fewer contaminants such as cellular debris and degradation products, and is more soluble, making it highly suitable for biological assays[292]. This method allows researchers to monitor venom composition with individual snails over time using mass spectrometry. Expanding sustainable bio-farming facilities has generated positive results[296]. Venom milking aligns with sustainable research practices by requiring fewer specimens and avoiding animal harm. Combining this method with venom duct transcriptome analysis of cDNA supports the conservation of cone snail populations and ensures continued progress in discovering novel pharmacological and potential drug candidates.
In essence, preserving the natural habitats of cone species is imperative for several critical reasons: (i) New cone snail species are constantly being discovered, and each species' unique pharmacological profile is vital to consider[297,298]. (ii) As outlined earlier, the crude venom of a cone snail, which can be ‘milked’ from the cone snail, is a much easier way to transport cone snail venom to different research facilities due to the current regulations surrounding individual cone snail peptides[1]. (iii) Sustainable bio-farming for this is likely the future, although there are limitations, too[299]. (iv) Despite the notable advances in the synthetic synthesis of cone snail peptides and cDNA venom duct transcriptome analysis, it remains helpful to separate crude venom using RP-HPLC as done by Yeung et al.[45,300], 2024 when they isolated Consomatins from C. geographus venom[45]. (v) In vitro folding of the chemically-synthesized peptides, where the disulfide pairings are correct, is not straightforward as the thermodynamically stable pairing is not necessarily the ones found in the native forms that are directly sourced from the cone snail venom[48].
The loss of cone snail habitats would significantly hinder these research processes, making discovering and applying novel and existing conotoxins more challenging. While synthetic approaches will likely play an ever-more critical role in the future, especially in the pharmaceutical industry, the combined use of crude venom and synthetic methods remains indispensable in conotoxin research[292]. Preserving cone snail habitats is not just an ethical obligation but a cost-effective and practical strategy for advancing biomedical research and ensuring sustainable access to this invaluable natural resource for future generations.
6 CONCLUSION
This review showcases the significant potential of conotoxins in neuropharmacology research and drug development. Despite decades of research, these peptides from marine cone snails remain a promising and unexplored area, with their diversity growing by about 50% in the last ten years[3,18,301].
The conotoxins’ exceptional specificity and potency make them invaluable tools for modulating ion channels, ligand-gated channels, receptors, and proteins involved in numerous physiological activities[102,302]. They hold significant potential in the search for new pharmaceutical compounds. The transition of conotoxins from natural venom elements to possible therapeutics showcases the efficacy and promise of natural compounds in pharmaceutical research[16,24].
Conotoxins have profoundly impacted pharmacological research, providing insights into the complex mechanisms of physiological and pathological processes. The FDA-approved ω-MVIIA for severe chronic pain management exemplifies the medicinal potential of venom-derived compounds[248]. This landmark achievement underlines the capacity of natural molecules to address unmet medical needs, particularly in areas where conventional therapies have fallen short or failed. However, transitioning from venom to medication presents challenges like structural complexity, stability, bioavailability, and possible immunogenicity. Modern technologies and innovative approaches, including enhanced specialised human receptor model testing, are crucial for progress[117]. This testing has significantly advanced our understanding of conotoxin pharmacology, and their interaction with human biological systems, essential for developing safer, more efficient drugs.
The discoveries of novel conotoxin classes, such as consomatins in 2022 and con-insulins in 2015, further show the growing evolution of this field. Notably, both of these classes have been isolated from C. geographus, a species that already had been studied for over six decades, and yet such discoveries have led to developments like mini-Ins[6,8,178,193,246,267]. The advent of advanced transcriptome mining techniques such as ConusPipe and the burgeoning integration of AI and ML into conotoxins research, promises to accelerate the pace of discovery, unlocking a plethora of novel conotoxins classes with unprecedented speed and efficiency[280-283].
The future of conotoxins in pharmacology is bright, with potential applications in chronic pain, diabetes, cardiovascular diseases, and neurological disorders. Creating synthetic analogues and modified versions of conotoxins supports this progress, potentially leading to novel medications with enhanced effectiveness and fewer side effects, Leconotide and α-Mr1.1[S4Dap, C16Pen] being good examples[54,273,277,278].
Despite the challenges that remain, the remarkable progress made in understanding and harnessing the unique properties of conotoxins offers a compelling model for drug discovery. Their exceptional specificity, potency, and adaptability position them as invaluable tools for developing innovative therapies for a wide range of medical conditions.
Acknowledgements
Mok KH was funded through a European Union Horizon 2020 (EU) Research and Innovation Programme under the Marie Skłodowska-Curie Actions grant agreement (No.721906-TRACT).
Conflicts of Interest
The authors declared no conflict of interest.
Data Availability
Data sharing is not applicable to this review as no datasets were generated or analyzed during the current study.
Copyright Permissions
Copyright © 2025 The Author(s). Published by Innovation Forever Publishing Group Limited. This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.
Author Contribution
Mok KH, based upon the lab’s research in this area, conceived and suggested writing a comprehensive review manuscript. Ward AJ performed the literature search, data analysis and wrote the bulk of the text. Both Ward AJ and Mok KH made the final revision of the manuscript.
Abbreviation List
ADM, Adriamycin
AFP3, Alpha-fetoprotein domain 3
AI, Artificial intelligence
AMI, Acute myocardial infarction
AOO, Area of occupancy
AUC, Area under the curve
AUD, Alcohol use disorder
AUPR, Area under the precision-recall curve
Bax/Bcl-2, Apoptosis regulators
BeAtMuSiC, Binding affinity predication through mutational scanning
BLAST, Basic local alignment search tool
CaV, Voltage-gated calcium channels
CBA/Caj, Specific mouse straina
cDNA, Complementary DNA
CG_US, Coarse-grained umbrella sampling
CGRP, Calcitonin gene-related peptide
CGX, Contulakin-G
CIBP, Cancer-induced bone pain
CINs, Cholinergic interneurons
CIPN, Chemotherapy-induced peripheral neuropathy
CPA, Conditioned place aversion
CPP, Conditioned place preference
CRISPR, Clustered regularly interspaced short palindromic repeats
Cryo-EM, Cryo-electron microscopy
CSF, Cerebrospinal fluid
CSS, Collisional cross-section
Cys, Cysteine residues
Cα RMSD, Alpha carbon root mean square deviation
DA, Dopamine
DEGs, Differentially expressed genes
DEGs, Differentially expressed genes
DRG, dorsal root ganglia
EC50, Concentration producing half-maximal effect
ELPM, Elevated plus maze
EM, Energy minimization
EPM, Elevated plus maze
EPSC, Excitatory postsynaptic current
EtOH, Ethanol
EU, European union
F1 score, measure of a test’s accuracy
FDA, U.S. Food and drug administration
FnIII-1, Fibronectin type III domain 1
GPCR, G-protein-coupled receptor
HEL, Human embryonic lung fibroblasts
hIR, Human insulin receptor
HMMER, Hidden markov model-based search
IC50, Half-maximal inhibitory concentration
ICK, Inhibitory cystine knot
iLTD, Inhibitor long-term depression
IM, Intramuscular
IP, Intraperitoneal
IV, Intravenous
Ki, Inhibition constant
KO, Knockout
KV, Voltage-gated potassium channels
LD50, Medial lethal dose
LMA, Low locomotor activity
MCC, Matthews Correlation Coefficient
MD, Molecular Dynamics
MIC, Minimal inhibitory concentration
ML, Machine Learning
MMGBSA, Molecular mechanics generalized born surface area
MMPBSA, Molecular mechanics Poisson-Boltzmann surface area
MPAs, Marine protected areas
mRNA, Messenger RNA
MS/MS, Tandem mass spectrometry
MTD, Maximum tolerated dose
MWM, Morris water maze
NAc, Nucleus accumbens
nAChRs, Nicotinic acetylcholine receptors
NaV, Voltage-gated sodium channels
NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells
NGS, Next-generation sequencing
NMR, Nuclear magnetic resonance
NSCLC, Non-small cell lung carcinoma
NT, Neurotensin
NTSR, Neurotensin receptor isoform
OFT, Open field test
PACAP, Pituitary adenylate cyclase-activating polypeptide
p-AKT, Phosphorylated protein kinase B
PBS, Phosphate-buffered saline
PCA, Prinical component analysis
p-ERK, Phosphorylated extracellular signal-regulated kinase
pIC50, Negative logarithm of the IC50 value
PIP2, Phosphatidylinositol 4,5-biphosphate
p-JNK, Phosphorylated c-Jun N-terminal kinase
p-mTOR, Phosphorylated mammalian target of rapamycin
PPI, Protein-protein interaction
Prialt®, Synthetic ω-MVIIA
ProGen, Protein generative model
pSNL, Partial sciatic nerve ligation
p-STAT3, Phosphorylated signal transducer and activator of transcription 3
PTMs, Post-translation modifications
PWL, Paw withdrawal latency
PWT, Paw withdrawal threshold
qPCR, Quantitative polymerase chain reaction
qRT-PCR, Quantitative real-time polymerase chain reaction
RMSD, Root mean square deviation
RP-HPLC, Reverse-phase high-performance liquid chromatography
SAR, Structure-activity relationship
SEM, Standard error of the mean
SGF, Simulated gastric fluid
SIF, Simulated intestinal fluid
SMOTE, Synthetic minority over-sampling technique
SSTRs, Somatostatin receptors
SVM, Support vector machine
TEVC, Two-electrode voltage clamp
TI, Therapeutic index
TNBC, Triple-negative breast cancer
TTX, Tetrodotoxin
TUD, Tobacco use disorder
UniProt, Universal protein resource
VGCCs, Voltage-gated calcium channels
VGSCs, Voltage-gated sodium channels
VTA, Ventral tegmental area
WAE, Wasserstein autoencoder
WT, Wild-type
μIR, Micro insulin receptor
References
[1] Bjørn-Yoshimoto WE, Ramiro IBL, Yandell M et al. Curses or Cures: A Review of the Numerous Benefits Versus the Biosecurity Concerns of Conotoxin Research. Biomedicines, 2020; 8.[DOI]
[2] KOHN AJ, NISHI M, PERNET B. Snail spears and scimitars: a character analysis of Conus radular teeth. J Mollus Stud, 1999, 65: 461-481.[DOI]
[3] Jin A-H, Muttenthaler M, Dutertre S et al. Conotoxins: Chemistry and Biology. Chem Rev, 2019; 119: 11510-11549.[DOI]
[4] Essack M, Bajic VB, Archer JAC. Conotoxins that confer therapeutic possibilities. Mar Drugs, 2012; 10: 1244-1265.[DOI]
[5] Becker S, Terlau H. Toxins from cone snails: properties, applications and biotechnological production. Appl Microbiol Biot, 2008; 79: 1-9.[DOI]
[6] Whyte JM, Endean R. Pharmacological investigation of the venoms of the marine snails Conus textile and Conus geographus. Toxicon, 1962; 1: 25-31.[DOI]
[7] Endean R, Gyr P, Parish G. Pharmacology of the venom of the gastropod Conus magus. Toxicon, 1974; 12: 117-129.[DOI]
[8] Endean R, Parish G, Gyr P. Pharmacology of the venom of Conus geographus. Toxicon, 1974; 12: 131-138.[DOI]
[9] Endean R, Williams H, Gyr P et al. Some effects on muscle and nerve of crude venom from the gastropod Conus striatus. Toxicon, 1976; 14: 267-272.[DOI]
[10] Endean R, Surridge J, Gyr P. Some effects of crude venom from the cones Conus striatus and Conus magus on isolated guinea-pig atria. Toxicon, 1977; 15: 369-374.[DOI]
[11] Endean R, Gyr P, Surridge J. The pharmacological actions on guinea-pig ileum of crude venoms from the marine gastropods Conus striatus and Conus magus. Toxicon, 1977; 15: 327-337.[DOI]
[12] Endean R, Gyr P, Surridge J. The effects of crude venoms of Conus magus and Conus striatus on the contractile response and electrical activity of guinea-pig cardiac musculature. Toxicon, 1979; 17: 381-395.[DOI]
[13] Pardi A, Galdes A, Florance J et al. Solution structures of alpha-conotoxin G1 determined by two-dimensional NMR spectroscopy. Biochemistry, 1989; 28: 5494-5501.[DOI]
[14] Olivera BM, Gray WR, Zeikus R et al. Peptide Neurotoxins from Fish-Hunting Cone Snails. Science, 1985; 230: 1338-1343.[DOI]
[15] Staats PS, Yearwood T, Charapata SG et al. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. JAMA, 2004; 291: 63-70.[DOI]
[16] Safavi-Hemami H, Brogan SE, Olivera BM. Pain therapeutics from cone snail venoms: From Ziconotide to novel non-opioid pathways. J Proteomics, 2019; 190: 12-20.[DOI]
[17] Lin J, Chen S, Butt UD et al. A comprehensive review on ziconotide. Heliyon, 2024; 10: e31105.[DOI]
[18] Spencer MR GM, Miniño AM. Drug Overdose Deaths in the United Sates, 2002-2022. NCHS Data Brief, no 491. Hyattsville, MD. 2024.[DOI]
[19] CDC NCfHS. Wide-ranging Online Data for Epidemiologic Research (WONDER). Atlanta, GA. 2021.
[20] Rubiu E, Restelli F, Nazzi V et al. A Benefit/Risk Assessment of Intrathecal Ziconotide in Chronic Pain: A Narrative Review. J Clin Med, 2024; 13: 1644.[DOI]
[21] Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev, 2004; 84: 41-68.[DOI]
[22] Olivera BM, Walker C, Cartier GE et al. Speciation of cone snails and interspecific hyperdivergence of their venom peptides. Potential evolutionary significance of introns. Ann N Y Acad Sci, 1999; 870: 223-237.[DOI]
[23] Prashanth JR, Brust A, Jin AH et al. Cone snail venomics: from novel biology to novel therapeutics. Future Med Chem, 2014; 6: 1659-1675.[DOI]
[24] Pennington MW, Czerwinski A, Norton RS. Peptide therapeutics from venom: Current status and potential. Bioorgan Med Chem, 2018; 26: 2738-2758.[DOI]
[25] Kaas Q, Westermann J-C, Craik DJ. Conopeptide characterization and classifications: an analysis using ConoServer. Toxicon, 2010; 55: 1491-1509.[DOI]
[26] Akondi KB, Muttenthaler M, Dutertre S et al. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem Rev, 2014; 114: 5815-5847.[DOI]
[27] Kaas Q, Yu R, Jin AH et al. ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res, 2012; 40: D325-330.[DOI]
[28] Robinson SD, Norton RS. Conotoxin gene superfamilies. Mar Drugs, 2014; 12: 6058-6101.[DOI]
[29] Ratibou Z, Inguimbert N, Dutertre S. Predatory and Defensive Strategies in Cone Snails. Toxins, 2024; 16: 94.[DOI]
[30] Gray WR, Luque A, Olivera BM et al. Peptide toxins from Conus geographus venom. J Biol Chem, 1981; 256: 4734-4740.[DOI]
[31] Wilhelm P, Luna-Ramirez K, Chin YK et al. Cysteine-Rich α-Conotoxin SII Displays Novel Interactions at the Muscle Nicotinic Acetylcholine Receptor. ACS Chem Neurosci, 2022; 13: 1245-1250.[DOI]
[32] Chen Z, Rogge G, Hague C et al. Subtype-selective noncompetitive or competitive inhibition of human alpha1-adrenergic receptors by rho-TIA. J Biol Chem, 2004; 279: 35326-35333.[DOI]
[33] Sharpe IA, Palant E, Schroeder CI et al. Inhibition of the norepinephrine transporter by the venom peptide chi-MrIA. Site of action, Na+ dependence, and structure-activity relationship. J Biol Chem, 2003; 278: 40317-40323.[DOI]
[34] Petrel C, Hocking HG, Reynaud M et al. Identification, structural and pharmacological characterization of τ-CnVA, a conopeptide that selectively interacts with somatostatin sst3 receptor. Biochem Pharmacol, 2013; 85: 1663-1671.[DOI]
[35] Safo P, Rosenbaum T, Shcherbatko A et al. Distinction among neuronal subtypes of voltage-activated sodium channels by mu-conotoxin PIIIA. J Neurosci, 2000; 20: 76-80.[DOI]
[36] Hasan MM, Starobova H, Mueller A et al. Subcutaneous ω-Conotoxins Alleviate Mechanical Pain in Rodent Models of Acute Peripheral Neuropathy. Mar Drugs, 2021; 19.[DOI]
[37] Kwon S, Bosmans F, Kaas Q et al. Efficient enzymatic cyclization of an inhibitory cystine knot-containing peptide. Biotechnol Bioeng, 2016; 113: 2202-2212.[DOI]
[38] Fainzilber M, Nakamura T, Lodder JC et al. gamma-Conotoxin-PnVIIA, a gamma-carboxyglutamate-containing peptide agonist of neuronal pacemaker cation currents. Biochemistry, 1998; 37: 1470-1477.[DOI]
[39] Jimenez EC, Shetty RP, Lirazan M et al. Novel excitatory Conus peptides define a new conotoxin superfamily. J Neurochem, 2003; 85: 610-621.[DOI]
[40] Wilson MJ, Yoshikami D, Azam L et al. μ-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. PNAS, 2011; 108: 10302-10307.[DOI]
[41] Jin AH, Dekan Z, Smout MJ et al. Conotoxin Φ-MiXXVIIA from the Superfamily G2 Employs a Novel Cysteine Framework that Mimics Granulin and Displays Anti-Apoptotic Activity. Angew Chem Int Ed Engl, 2017; 56: 14973-14976.[DOI]
[42] Christensen SB, Bandyopadhyay PK, Olivera BM et al. αS-conotoxin GVIIIB potently and selectively blocks α9α10 nicotinic acetylcholine receptors. Biochem Pharmacol, 2015; 96: 349-356.[DOI]
[43] Menting JG, Gajewiak J, MacRaild CA et al. A minimized human insulin-receptor-binding motif revealed in a Conus geographus venom insulin. Nat Struct Mol Biol, 2016; 23: 916-920.[DOI]
[44] Dutertre S, Croker D, Daly NL et al. Conopressin-T from Conus tulipa reveals an antagonist switch in vasopressin-like peptides. J Biol Chem, 2008; 283: 7100-7108.[DOI]
[45] Yeung HY, Ramiro IBL, Andersen DB et al. Fish-hunting cone snail disrupts prey's glucose homeostasis with weaponized mimetics of somatostatin and insulin. Nat Commun, 2024; 15: 6408.[DOI]
[46] Craig AG, Norberg T, Griffin D et al. Contulakin-G, an O-glycosylated invertebrate neurotensin. J Biol Chem, 1999; 274: 13752-13759.[DOI]
[47] Robinson SD, Safavi-Hemami H, McIntosh LD et al. Diversity of conotoxin gene superfamilies in the venomous snail, Conus victoriae. PLoS One, 2014; 9: e87648.[DOI]
[48] Bulaj G, Olivera BM. Folding of conotoxins: formation of the native disulfide bridges during chemical synthesis and biosynthesis of Conus peptides. Antioxid Redox Signal, 2008; 10: 141-155.[DOI]
[49] McIntosh JM, Yoshikami D, Mahe E et al. A nicotinic acetylcholine receptor ligand of unique specificity, alpha-conotoxin ImI. J Biol Chem, 1994; 269: 16733-16739.[DOI]
[50] Tabassum N, Tae HS, Jia X et al. Role of Cys(I)-Cys(III) Disulfide Bond on the Structure and Activity of α-Conotoxins at Human Neuronal Nicotinic Acetylcholine Receptors. ACS Omega, 2017; 2: 4621-4631.[DOI]
[51] Armishaw CJ, Banerjee J, Ganno ML et al. Discovery of novel antinociceptive α-conotoxin analogues from the direct in vivo screening of a synthetic mixture-based combinatorial library. Acs Comb Sci, 2013; 15: 153-161.[DOI]
[52] Mei D, Lin Z, Fu J et al. The use of α-conotoxin ImI to actualize the targeted delivery of paclitaxel micelles to α7 nAChR-overexpressing breast cancer. Biomaterials, 2015; 42: 52-65.[DOI]
[53] Mei D, Zhao L, Chen B et al. α-Conotoxin ImI-modified polymeric micelles as potential nanocarriers for targeted docetaxel delivery to α7-nAChR overexpressed non-small cell lung cancer. Drug Deliv, 2018; 25: 493-503.[DOI]
[54] Chen X, Zou Z, Li W et al. α-Conotoxin recombinant protein ImI-AFP3 efficiently inhibits the growth and migration of lung cancer cells. Protein Expr Purif, 2024; 215: 106405.[DOI]
[55] Lin B, Dong X, Wang Q et al. AFP-Inhibiting Fragments for Drug Deliv: The Promise and Challenges of Targeting Therapeutics to Cancers. Front Cell Dev Biol, 2021; 9.[DOI]
[56] Cartier GE, Yoshikami D, Gray WR et al. A new alpha-conotoxin which targets alpha3beta2 nicotinic acetylcholine receptors. J Biol Chem, 1996; 271: 7522-7528.[DOI]
[57] Tavazoie SF, Tavazoie MF, McIntosh JM et al. Differential block of nicotinic synapses on B versus C neurones in sympathetic ganglia of frog by alpha-conotoxins MII and ImI. Br J Pharmacol, 1997; 120: 995-1000.[DOI]
[58] Klink R, de Kerchove d'Exaerde A, Zoli M et al. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci, 2001; 21: 1452-1463.[DOI]
[59] Champtiaux N, Han ZY, Bessis A et al. Distribution and pharmacology of alpha 6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci, 2002; 22: 1208-1217.[DOI]
[60] Salminen O, Murphy KL, McIntosh JM et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol, 2004; 65: 1526-1535.[DOI]
[61] Zoli M, Moretti M, Zanardi A et al. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci, 2002; 22: 8785-8789.[DOI]
[62] McIntosh JM, Azam L, Staheli S et al. Analogs of α-Conotoxin MII Are Selective for α6-Containing Nicotinic Acetylcholine Receptors. Mol Pharmacol, 2004; 65: 944-952.[DOI]
[63] Jackson KJ, McIntosh JM, Brunzell DH et al. The role of alpha6-containing nicotinic acetylcholine receptors in nicotine reward and withdrawal. J Pharmacol Exp Ther, 2009; 331: 547-554.[DOI]
[64] Larsson A, Jerlhag E, Svensson L et al. Is an alpha-conotoxin MII-sensitive mechanism involved in the neurochemical, stimulatory, and rewarding effects of ethanol? Alcohol, 2004; 34: 239-250.[DOI]
[65] Wills L, Ables JL, Braunscheidel KM et al. Neurobiological Mechanisms of Nicotine Reward and Aversion. Pharmacol Rev, 2022; 74: 271-310.[DOI]
[66] Palkovits M, Brownstein MJ. Locus Coeruleus. In: Advances in Cellular Neurobiology. Elsevier, 1983; 81-103.[DOI]
[67] Sanjakdar SS, Maldoon PP, Marks MJ et al. Differential Roles of α6β2* and α4β2* Neuronal Nicotinic Receptors in Nicotine- and Cocaine-Conditioned Reward in Mice. Neuropsychopharmacology, 2015; 40: 350-360.[DOI]
[68] Wadsworth HA, Anderson EQ, Williams BM et al. Role of α6-Nicotinic Receptors in Alcohol-Induced GABAergic Synaptic Transmission and Plasticity to Cholinergic Interneurons in the Nucleus Accumbens. Mol Neurobiol, 2023; 60: 3113-3129.[DOI]
[69] Koob GF. Alcoholism: allostasis and beyond. Alcohol Clin Exp Res, 2003; 27: 232-243.[DOI]
[70] Xu X, Bishop EE, Kennedy SM et al. Annual healthcare spending attributable to cigarette smoking: an update. Am J Prev Med, 2015; 48: 326-333.[DOI]
[71] Powers MS, Broderick HJ, Drenan RM et al. Nicotinic acetylcholine receptors containing α6 subunits contribute to alcohol reward-related behaviours. Genes Brain Behav, 2013; 12: 543-553.[DOI]
[72] Tosti E, Boni R, Gallo A. µ-Conotoxins Modulating Sodium Currents in Pain Perception and Transmission: A Therapeutic Potential. Mar Drugs, 2017; 15.[DOI]
[73] Norton RS. µ-Conotoxins as Leads in the Development of New Analgesics. Molecules, 2010; 15: 2825-2844.[DOI]
[74] Wang H, Huang J, Zang J et al. Drug discovery targeting Nav1.8: Structural insights and therapeutic potential. Curr Opin Chem Biol, 2024; 83: 102538.[DOI]
[75] McMahon KL, Vetter I, Schroeder CI. Voltage-Gated Sodium Channel Inhibition by µ-Conotoxins. Toxins, 2024; 16: 55.[DOI]
[76] Khoo KK, Feng ZP, Smith BJ et al. Structure of the analgesic mu-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry, 2009; 48: 1210-1219.[DOI]
[77] Khoo KK, Gupta K, Green BR et al. Distinct disulfide isomers of μ-conotoxins KIIIA and KIIIB block voltage-gated sodium channels. Biochemistry, 2012; 51: 9826-9835.[DOI]
[78] Tran HNT, McMahon KL, Deuis JR et al. Structural and functional insights into the inhibition of human voltage-gated sodium channels by μ-conotoxin KIIIA disulfide isomers. J Biol Chem, 2022; 298: 101728.[DOI]
[79] Van Der Haegen A, Peigneur S, Tytgat J. Importance of position 8 in μ-conotoxin KIIIA for voltage-gated sodium channel selectivity. Febs j, 2011; 278: 3408-3418.[DOI]
[80] Han P, Wang K, Dai X et al. The Role of Individual Disulfide Bonds of μ-Conotoxin GIIIA in the Inhibition of Na(V)1.4. Mar Drugs, 2016; 14.[DOI]
[81] Leipold E, Ullrich F, Thiele M et al. Subtype-specific block of voltage-gated K(+) channels by μ-conopeptides. Biochem Biophys Res Commun, 2017; 482: 1135-1140.[DOI]
[82] Nakamura M, Ishida Y, Kohno T et al. Synthesis of [Cys(5)]mu-conotoxin GIIIA and its derivatives as a probe of Na(+) channel analysis. Biochem Biophys Res Commun, 2001; 283: 374-378.[DOI]
[83] Patel D, Mahdavi S, Kuyucak S. Computational Study of Binding of μ-Conotoxin GIIIA to Bacterial Sodium Channels NaVAb and NaVRh. Biochemistry, 2016; 55: 1929-1938.[DOI]
[84] Zou X, Zhang Z, Lu H et al. Functional effects of drugs and toxins interacting with Na(V)1.4. Front Pharmacol, 2024; 15: 1378315.[DOI]
[85] Norton RS, Pallaghy PK. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon, 1998; 36: 1573-1583.[DOI]
[86] Olivera BM, McIntosh JM, Cruz LJ et al. Purification and sequence of a presynaptic peptide toxin from Conus geographus venom. Biochemistry, 1984; 23: 5087-5090.[DOI]
[87] Lew MJ, Flinn JP, Pallaghy PK et al. Structure-function relationships of omega-conotoxin GVIA. Synthesis, structure, calcium channel binding, and functional assay of alanine-substituted analogues. J Biol Chem, 1997; 272: 12014-12023.[DOI]
[88] Favreau P, Gilles N, Lamthanh H et al. A new omega-conotoxin that targets N-type voltage-sensitive calcium channels with unusual specificity. Biochemistry, 2001; 40: 14567-14575.[DOI]
[89] Lewis RJ, Nielsen KJ, Craik DJ et al. Novel omega-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J Biol Chem, 2000; 275: 35335-35344.[DOI]
[90] Schroeder CI, Lewis RJ. ω-Conotoxins GVIA, MVIIA and CVID: SAR and Clinical Potential. Mar Drugs, 2006; 4: 193-214.[DOI]
[91] Lewis RJ, Garcia ML. Therapeutic potential of venom peptides. Nat Rev Drug Discov, 2003; 2: 790-802.[DOI]
[92] Bowersox S, Valentino K, Luther R. Neuronal voltage-sensitive calcium channels. Drug News Perspects, 1994; 7: 261-261.
[93] Valentino K, Newcomb R, Gadbois T et al. A selective N-type calcium channel antagonist protects against neuronal loss after global cerebral ischemia. PNAS, 1993; 90: 7894-7897.[DOI]
[94] Janes RW. alpha-Conotoxins as selective probes for nicotinic acetylcholine receptor subclasses. Curr Opin Pharmacol, 2005; 5: 280-292.[DOI]
[95] Dutton JL, Craik DJ. alpha-Conotoxins: nicotinic acetylcholine receptor antagonists as pharmacological tools and potential drug leads. Curr Med Chem, 2001; 8: 327-344.[DOI]
[96] Giribaldi J, Dutertre S. α-Conotoxins to explore the molecular, physiological and pathophysiological functions of neuronal nicotinic acetylcholine receptors. Neurosci Lett, 2018; 679: 24-34.[DOI]
[97] Cho JH, Mok KH, Olivera BM et al. Nuclear magnetic resonance solution conformation of alpha-conotoxin AuIB, an alpha(3)beta(4) subtype-selective neuronal nicotinic acetylcholine receptor antagonist. J Biol Chem, 2000; 275: 8680-8685.[DOI]
[98] Talley TT, Olivera BM, Han KH et al. Alpha-conotoxin OmIA is a potent ligand for the acetylcholine-binding protein as well as alpha3beta2 and alpha7 nicotinic acetylcholine receptors. J Biol Chem, 2006; 281: 24678-24686.[DOI]
[99] Chi SW, Park KH, Suk JE et al. Solution conformation of alphaA-conotoxin EIVA, a potent neuromuscular nicotinic acetylcholine receptor antagonist from Conus ermineus. J Biol Chem, 2003; 278: 42208-42213.[DOI]
[100] Chi SW, Kim DH, Olivera BM et al. Solution conformation of alpha-conotoxin GIC, a novel potent antagonist of alpha3beta2 nicotinic acetylcholine receptors. Biochem J, 2004; 380: 347-352.[DOI]
[101] Abraham N, Healy M, Ragnarsson L et al. Structural mechanisms for α-conotoxin activity at the human α3β4 nicotinic acetylcholine receptor. Sci Rep, 2017; 7: 45466.[DOI]
[102] Champtiaux N, Gotti C, Cordero-Erausquin M et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci, 2003; 23: 7820-7829.[DOI]
[103] Hansen SB, Sulzenbacher G, Huxford T et al. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J, 2005; 24: 3635-3646.[DOI]
[104] Mok KH, Han KH. NMR solution conformation of an antitoxic analogue of alpha-conotoxin GI: identification of a common nicotinic acetylcholine receptor alpha 1-subunit binding surface for small ligands and alpha-conotoxins. Biochemistry, 1999; 38: 11895-11904.[DOI]
[105] Park KH, Suk JE, Jacobsen R et al. Solution conformation of alpha-conotoxin EI, a neuromuscular toxin specific for the alpha 1/delta subunit interface of torpedo nicotinic acetylcholine receptor. J Biol Chem, 2001; 276: 49028-49033.[DOI]
[106] Chi SW, Lee SH, Kim DH et al. Solution structure of alpha-conotoxin PIA, a novel antagonist of alpha6 subunit containing nicotinic acetylcholine receptors. Biochem Biophys Res Commun, 2005; 338: 1990-1997.[DOI]
[107] Chi SW, Kim DH, Olivera BM et al. NMR structure determination of alpha-conotoxin BuIA, a novel neuronal nicotinic acetylcholine receptor antagonist with an unusual 4/4 disulfide scaffold. Biochem Biophys Res Commun, 2006; 349: 1228-1234.[DOI]
[108] Han KH, Hwang KJ, Kim SM et al. NMR structure determination of a novel conotoxin, [Pro 7,13] alpha A-conotoxin PIVA. Biochemistry, 1997; 36: 1669-1677.[DOI]
[109] Ma X, Huang Q, Yu S et al. The 3/4- and 3/6-Subfamily Variants of α-Conotoxins GI and MI Exhibit Potent Inhibitory Activity against Muscular Nicotinic Acetylcholine Receptors. Mar Drugs, 2021; 19.[DOI]
[110] Johnson DS, Martinez J, Elgoyhen AB et al. alpha-Conotoxin ImI exhibits subtype-specific nicotinic acetylcholine receptor blockade: preferential inhibition of homomeric alpha 7 and alpha 9 receptors. Mol Pharmacol, 1995; 48: 194-199.
[111] Ning J, Li R, Ren J et al. Alanine-Scanning Mutagenesis of α-Conotoxin GI Reveals the Residues Crucial for Activity at the Muscle Acetylcholine Receptor. Mar Drugs, 2018; 16.[DOI]
[112] Jacobsen RB, DelaCruz RG, Grose JH et al. Critical residues influence the affinity and selectivity of alpha-conotoxin MI for nicotinic acetylcholine receptors. Biochemistry, 1999; 38: 13310-13315.[DOI]
[113] Myers RA, Zafaralla GC, Gray WR et al. alpha-Conotoxins, small peptide probes of nicotinic acetylcholine receptors. Biochemistry, 1991; 30: 9370-9377.[DOI]
[114] Favreau P, Krimm I, Le Gall F et al. Biochemical characterization and nuclear magnetic resonance structure of novel alpha-conotoxins isolated from the venom of Conus consors. Biochemistry, 1999; 38: 6317-6326.[DOI]
[115] López-Vera E, Aguilar MB, Schiavon E et al. Novel alpha-conotoxins from Conus spurius and the alpha-conotoxin EI share high-affinity potentiation and low-affinity inhibition of nicotinic acetylcholine receptors. Febs j, 2007; 274: 3972-3985.[DOI]
[116] Ning J, Ren J, Xiong Y et al. Identification of Crucial Residues in α-Conotoxin EI Inhibiting Muscle Nicotinic Acetylcholine Receptor. Toxins (Basel), 2019; 11.[DOI]
[117] Hone AJ, Kaas Q, Kearns I et al. Computational and Functional Mapping of Human and Rat α6β4 Nicotinic Acetylcholine Receptors Reveals Species-Specific Ligand-Binding Motifs. J Med Chem, 2021; 64: 1685-1700.[DOI]
[118] Pucci L, Grazioso G, Dallanoce C et al. Engineering of α-conotoxin MII-derived peptides with increased selectivity for native α6β2* nicotinic acetylcholine receptors. Faseb j, 2011; 25: 3775-3789.[DOI]
[119] Beissner M, Dutertre S, Schemm R et al. Efficient binding of 4/7 α-conotoxins to nicotinic α4β2 receptors is prevented by Arg185 and Pro195 in the α4 subunit. Mol Pharmacol, 2012; 82: 711-718.[DOI]
[120] Dowell C, Olivera BM, Garrett JE et al. Alpha-conotoxin PIA is selective for alpha6 subunit-containing nicotinic acetylcholine receptors. J Neurosci, 2003; 23: 8445-8452.[DOI]
[121] Leffler AE, Kuryatov A, Zebroski HA et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. PNAS, 2017; 114: E8100-e8109.[DOI]
[122] Nicke A, Loughnan ML, Millard EL et al. Isolation, structure, and activity of GID, a novel alpha 4/7-conotoxin with an extended N-terminal sequence. J Biol Chem, 2003; 278: 3137-3144.[DOI]
[123] McIntosh JM, Dowell C, Watkins M et al. Alpha-conotoxin GIC from Conus geographus, a novel peptide antagonist of nicotinic acetylcholine receptors. J Biol Chem, 2002; 277: 33610-33615.[DOI]
[124] Souf YM, Lokaj G, Kuruva V et al. Synthesis and Biological Activity of Novel α-Conotoxins Derived from Endemic Polynesian Cone Snails. Mar Drugs, 2023; 21: 356.[DOI]
[125] Armishaw C, Jensen AA, Balle T et al. Rational design of alpha-conotoxin analogues targeting alpha7 nicotinic acetylcholine receptors: improved antagonistic activity by incorporation of proline derivatives. J Biol Chem, 2009; 284: 9498-9512.[DOI]
[126] Fainzilber M, Hasson A, Oren R et al. New mollusc-specific alpha-conotoxins block Aplysia neuronal acetylcholine receptors. Biochemistry, 1994; 33: 9523-9529.[DOI]
[127] Ho TNT, Lee HS, Swaminathan S et al. Posttranslational modifications of α-conotoxins: sulfotyrosine and C-terminal amidation stabilise structures and increase acetylcholine receptor binding. RSC Med Chem, 2021; 12: 1574-1584. [DOI]
[128] Nicke A, Samochocki M, Loughnan ML et al. Alpha-conotoxins EpI and AuIB switch subtype selectivity and activity in native versus recombinant nicotinic acetylcholine receptors. FEBS Lett, 2003; 554: 219-223.[DOI]
[129] Luo S, Kulak JM, Cartier GE et al. alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J Neurosci, 1998; 18: 8571-8579.[DOI]
[130] Armishaw CJ, Jensen AA, Balle LD et al. Improving the stability of α-conotoxin AuIB through N-to-C cyclization: the effect of linker length on stability and activity at nicotinic acetylcholine receptors. Antioxid Redox Signal, 2011; 14: 65-76.[DOI]
[131] Nevin ST, Clark RJ, Klimis H et al. Are alpha9alpha10 nicotinic acetylcholine receptors a pain target for alpha-conotoxins? Mol Pharmacol, 2007; 72: 1406-1410.[DOI]
[132] Ren J, Zhu X, Xu P et al. d-Amino Acid Substitution of α-Conotoxin RgIA Identifies its Critical Residues and Improves the Enzymatic Stability. Mar Drugs, 2019; 17.[DOI]
[133] Zheng N, Christensen SB, Dowell C et al. Discovery of Methylene Thioacetal-Incorporated α-RgIA Analogues as Potent and Stable Antagonists of the Human α9α10 Nicotinic Acetylcholine Receptor for the Treatment of Neuropathic Pain. J Med Chem, 2021; 64: 9513-9524.[DOI]
[134] Ellison M, Feng ZP, Park AJ et al. Alpha-RgIA, a novel conotoxin that blocks the alpha9alpha10 nAChR: structure and identification of key receptor-binding residues. J Mol Biol, 2008; 377: 1216-1227.[DOI]
[135] Vincler M, Wittenauer S, Parker R et al. Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors. PNAS, 2006; 103: 17880-17884.[DOI]
[136] Ellison M, Haberlandt C, Gomez-Casati ME et al. Alpha-RgIA: a novel conotoxin that specifically and potently blocks the alpha9alpha10 nAChR. Biochemistry, 2006; 45: 1511-1517.[DOI]
[137] Ellison M, Gao F, Wang HL et al. Alpha-conotoxins ImI and ImII target distinct regions of the human alpha7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry, 2004; 43: 16019-16026.[DOI]
[138] Loughnan ML, Nicke A, Jones A et al. Chemical and functional identification and characterization of novel sulfated alpha-conotoxins from the cone snail Conus anemone. J Med Chem, 2004; 47: 1234-1241.[DOI]
[139] Di Lascio S, Fornasari D, Benfante R. The Human-Restricted Isoform of the α7 nAChR, CHRFAM7A: A Double-Edged Sword in Neurological and Inflammatory Disorders. Int J Mol Sci, 2022; 23: 3463.[DOI]
[140] Morrison KL, Weiss GA. Combinatorial alanine-scanning. Curr Opin Chem Biol, 2001; 5: 302-307.[DOI]
[141] Wang S, Zhu X, Zhangsun M et al. Engineered Conotoxin Differentially Blocks and Discriminates Rat and Human α7 Nicotinic Acetylcholine Receptors. J Med Chem, 2021; 64: 5620-5631.[DOI]
[142] Leiser SC, Bowlby MR, Comery TA et al. A cog in cognition: how the alpha 7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol Ther, 2009; 122: 302-311.[DOI]
[143] Vallés AS, Barrantes FJ. Nicotinic Acetylcholine Receptor Dysfunction in Addiction and in Some Neurodegenerative and Neuropsychiatric Diseases. Cells, 2023; 12.[DOI]
[144] Freedman R, Coon H, Myles-Worsley M et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. PNAS, 1997; 94: 587-592.[DOI]
[145] Shan H, Wang N, Gao X et al. Fluorescent α-Conotoxin [Q1G, ΔR14]LvIB Identifies the Distribution of α7 Nicotinic Acetylcholine Receptor in the Rat Brain. Mar Drugs, 2024; 22.[DOI]
[146] Xu C, Wang N, Ma T et al. The α3β4 nAChR tissue distribution identified by fluorescent α-conotoxin [D11A]LvIA. Int J Biol Macromol, 2024; 281: 136220.[DOI]
[147] Qian J, Liu YQ, Sun ZH et al. Identification of nicotinic acetylcholine receptor subunits in different lung cancer cell lines and the inhibitory effect of alpha-conotoxin TxID on lung cancer cell growth. Eur J Pharmacol, 2019; 865: 172674.[DOI]
[148] You S, Li X, Xiong J et al. α-Conotoxin TxIB: A Uniquely Selective Ligand for α6/α3β2β3 Nicotinic Acetylcholine Receptor Attenuates Nicotine-Induced Conditioned Place Preference in Mice. Mar Drugs, 2019; 17.[DOI]
[149] Li X, Xiong J, Zhang B et al. α-Conotoxin TxIB Inhibits Development of Morphine-Induced Conditioned Place Preference in Mice via Blocking α6β2* Nicotinic Acetylcholine Receptors. Front Pharmacol, 2021; 12: 772990.[DOI]
[150] Mao K, Li X, Chen Z et al. α-Conotoxin TxIB Improved Behavioral Abnormality and Changed Gene Expression in Zebrafish (Danio rerio) Induced by Alcohol Withdrawal. Front Pharmacol, 2022; 13: 802917.[DOI]
[151] Wang M, Liao Z, Zhangsun D et al. Engineering Enhanced Antimicrobial Properties in α-Conotoxin RgIA through D-Type Amino Acid Substitution and Incorporation of Lysine and Leucine Residues. Molecules, 2024; 29.[DOI]
[152] Azam L, Christensen SB, Riaz Z et al. α9-Containing Nicotinic Acetylcholine Receptors Are Required for RgIA-5474 Attenuation of Chemotherapy-Induced Neuropathic Pain. ACS Pharmacol Transl Sci, 2024; 7: 3935-3944.[DOI]
[153] Li T, Tae HS, Liang J et al. Rational Design of Potent α-Conotoxin PeIA Analogues with Non-Natural Amino Acids for the Inhibition of Human α9α10 Nicotinic Acetylcholine Receptors. Mar Drugs, 2024; 22.[DOI]
[154] Liang J, Tae HS, Zhao Z et al. Mechanism of Action and Structure-Activity Relationship of α-Conotoxin Mr1.1 at the Human α9α10 Nicotinic Acetylcholine Receptor. J Med Chem, 2022; 65: 16204-16217.[DOI]
[155] Li T, Tae HS, Chen S et al. Development of an Intravenously Stable Disulfide-Rich Peptide for the Treatment of Chemotherapy-Induced Neuropathic Pain. J Med Chem, 2024; 67: 18741-18752.[DOI]
[156] Li X, Tae HS, Chen S et al. Dual Antagonism of α9α10 nAChR and GABA(B) Receptor-Coupled Ca(V)2.2 Channels by an Analgesic αO-Conotoxin Analogue. J Med Chem, 2024; 67: 971-987.[DOI]
[157] Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annu Rev Neurosci, 1987; 10: 633-693.[DOI]
[158] Zamponi GW. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov, 2016; 15: 19-34.[DOI]
[159] Kaas Q, Westermann JC, Halai R et al. ConoServer, a database for conopeptide sequences and structures. Bioinformatics, 2008; 24: 445-446.[DOI]
[160] Monje VD, Haack JA, Naisbitt SR et al. A new Conus peptide ligand for Ca channel subtypes. Neuropharmacology, 1993; 32: 1141-1149.[DOI]
[161] Wen L, Yang S, Qiao H et al. SO-3, a new O-superfamily conopeptide derived from Conus striatus, selectively inhibits N-type calcium currents in cultured hippocampal neurons. Br J Pharmacol, 2005; 145: 728-739.[DOI]
[162] Wang F, Yan Z, Liu Z et al. Molecular basis of toxicity of N-type calcium channel inhibitor MVIIA. Neuropharmacology, 2016; 101: 137-145.[DOI]
[163] Berecki G, Motin L, Haythornthwaite A et al. Analgesic (omega)-conotoxins CVIE and CVIF selectively and voltage-dependently block recombinant and native N-type calcium channels. Mol Pharmacol, 2010; 77: 139-148.[DOI]
[164] Mould J, Yasuda T, Schroeder CI et al. The alpha2delta auxiliary subunit reduces affinity of omega-conotoxins for recombinant N-type (Cav2.2) calcium channels. J Biol Chem, 2004; 279: 34705-34714.[DOI]
[165] Berecki G, Daly NL, Huang YH et al. Effects of arginine 10 to lysine substitution on ω-conotoxin CVIE and CVIF block of Cav2.2 channels. Br J Pharmacol, 2014; 171: 3313-3327.[DOI]
[166] Kits KS, Lodder JC, van der Schors RC et al. Novel omega-conotoxins block dihydropyridine-insensitive high voltage-activated calcium channels in molluscan neurons. J Neurochem, 1996; 67: 2155-2163.[DOI]
[167] Fainzilber M, Lodder JC, Van der Schors RC et al. A novel hydrophobic ω-conotoxin blocks molluscan dihydropyridine-sensitive calcium channels. Biochemistry, 1996; 35: 8748-8752.[DOI]
[168] Sousa SR, McArthur JR, Brust A et al. Novel analgesic ??-conotoxins from the vermivorous cone snail Conus moncuri provide new insights into the evolution of conopeptides. Sci Rep, 2018; 8: 13397.[DOI]
[169] Bernáldez J, Román-González SA, Martínez O et al. A Conus regularis conotoxin with a novel eight-cysteine framework inhibits CaV2.2 channels and displays an anti-nociceptive activity. Mar Drugs, 2013; 11: 1188-1202.[DOI]
[170] Armishaw JC, Alewood FP. Conotoxins as Research Tools and Drug Leads. Curr Protein Pept Sc, 2005; 6: 221-240.[DOI]
[171] Pallaghy PK, Nielsen KJ, Craik DJ et al. A common structural motif incorporating a cystine knot and a triple-stranded beta-sheet in toxic and inhibitory polypeptides. Protein Sci, 1994; 3: 1833-1839.[DOI]
[172] Kolosov A, Aurini L, Williams ED et al. Intravenous injection of leconotide, an omega conotoxin: synergistic antihyperalgesic effects with morphine in a rat model of bone cancer pain. Pain Med, 2011; 12: 923-941.[DOI]
[173] Sanford M. Intrathecal ziconotide: a review of its use in patients with chronic pain refractory to other systemic or intrathecal analgesics. CNS Drugs, 2013; 27: 989-1002.[DOI]
[174] Jayamanne A, Jeong HJ, Schroeder CI et al. Spinal actions of ω-conotoxins, CVID, MVIIA and related peptides in a rat neuropathic pain model. Br J Pharmacol, 2013; 170: 245-254.[DOI]
[175] Chen J, Liu X, Yu S et al. A novel ??-conotoxin Bu8 inhibiting N-type voltage-gated calcium channels displays potent analgesic activity. Acta Pharm Sin B, 2021; 11: 2685-2693.[DOI]
[176] Nielsen KJ, Adams D, Thomas L et al. Structure-activity relationships of ω-conotoxins MVIIA, MVIIC and 14 loop splice hybrids at N and P/Q-type calcium channels. J Mol Biol, 1999; 289: 1405-1421.[DOI]
[177] Nielsen KJ, Adams DA, Alewood PF et al. Effects of chirality at Tyr13 on the structure-activity relationships of omega-conotoxins from Conus magus. Biochemistry, 1999; 38: 6741-6751.[DOI]
[178] Safavi-Hemami H, Gajewiak J, Karanth S et al. Specialized insulin is used for chemical warfare by fish-hunting cone snails. PNAS, 2015; 112: 1743-1748.[DOI]
[179] Ahorukomeye P, Disotuar MM, Gajewiak J et al. Fish-hunting cone snail venoms are a rich source of minimized ligands of the vertebrate insulin receptor. eLife, 2019; 8.[DOI]
[180] Dutt M, Dutertre S, Jin AH et al. Venomics Reveals Venom Complexity of the Piscivorous Cone Snail, Conus tulipa. Mar Drugs, 2019; 17.[DOI]
[181] Safavi-Hemami H, Lu A, Li Q et al. Venom Insulins of Cone Snails Diversify Rapidly and Track Prey Taxa. Mol Biol Evol, 2016; 33: 2924-2934.[DOI]
[182] Guo Q, Huang M, Li M et al. Diversity and Evolutionary Analysis of Venom Insulin Derived from Cone Snails. Toxins, 2024; 16: 34.[DOI]
[183] Shabanpoor F, Separovic F, Wade JD. The human insulin superfamily of polypeptide hormones. Vitam Horm, 2009; 80: 1-31.[DOI]
[184] Weiss MA, Lawrence MC. A thing of beauty: Structure and function of insulin’s “aromatic triplet”. Diabetes Obes Metab, 2018; 20 Suppl 2: 51-63.[DOI]
[185] Harding MM, Hodgkin DC, Kennedy AF et al. The crystal structure of insulin. II. An investigation of rhombohedral zinc insulin crystals and a report of other crystalline forms. J Mol Biol, 1966; 16: 212-226.[DOI]
[186] Jiráček J, Žáková L. From venom peptides to a potential diabetes treatment. Elife, 2019; 8.[DOI]
[187] Gorai B, Vashisth H. Structures and interactions of insulin-like peptides from cone snail venom. Proteins, 2022; 90: 680-690.[DOI]
[188] Catterall WA. Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev, 1992; 72: S15-48.[DOI]
[189] Lewis RJ, Dutertre S, Vetter I et al. Conus venom peptide pharmacology. Pharmacol Rev, 2012; 64: 259-298.[DOI]
[190] Becker S, Prusak-Sochaczewski E, Zamponi G et al. Action of derivatives of mu-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry, 1992; 31: 8229-8238.[DOI]
[191] Zhang MM, Fiedler B, Green BR et al. Structural and functional diversities among mu-conotoxins targeting TTX-resistant sodium channels. Biochemistry, 2006; 45: 3723-3732.[DOI]
[192] Favreau P, Benoit E, Hocking HG et al. A novel µ-conopeptide, CnIIIC, exerts potent and preferential inhibition of NaV1.2/1.4 channels and blocks neuronal nicotinic acetylcholine receptors. Br J Pharmacol, 2012; 166: 1654-1668.[DOI]
[193] Cruz LJ, Gray WR, Olivera BM et al. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J Biol Chem, 1985; 260: 9280-9288.[DOI]
[194] Harvey PJ, Kurniawan ND, Finol-Urdaneta RK et al. NMR Structure of μ-Conotoxin GIIIC: Leucine 18 Induces Local Repacking of the N-Terminus Resulting in Reduced Na(V) Channel Potency. Molecules, 2018; 23.[DOI]
[195] Walewska A, Skalicky JJ, Davis DR et al. NMR-based mapping of disulfide bridges in cysteine-rich peptides: application to the mu-conotoxin SxIIIA. J Am Chem Soc, 2008; 130: 14280-14286.[DOI]
[196] McMahon KL, Tran HNT, Deuis JR et al. Discovery, Pharmacological Characterisation and NMR Structure of the Novel µ-Conotoxin SxIIIC, a Potent and Irreversible Na(V) Channel Inhibitor. Biomedicines, 2020; 8.[DOI]
[197] Lewis RJ, Schroeder CI, Ekberg J et al. Isolation and structure-activity of mu-conotoxin TIIIA, a potent inhibitor of tetrodotoxin-sensitive voltage-gated sodium channels. Mol Pharmacol, 2007; 71: 676-685.[DOI]
[198] Yao S, Zhang MM, Yoshikami D et al. Structure, dynamics, and selectivity of the sodium channel blocker mu-conotoxin SIIIA. Biochemistry, 2008; 47: 10940-10949.[DOI]
[199] Schroeder CI, Ekberg J, Nielsen KJ et al. Neuronally micro-conotoxins from Conus striatus utilize an alpha-helical motif to target mammalian sodium channels. J Biol Chem, 2008; 283: 21621-21628.[DOI]
[200] Leipold E, Markgraf R, Miloslavina A et al. Molecular determinants for the subtype specificity of μ-conotoxin SIIIA targeting neuronal voltage-gated sodium channels. Neuropharmacology, 2011; 61: 105-111.[DOI]
[201] Wilson MJ, Zhang MM, Gajewiak J et al. Α- and β-subunit composition of voltage-gated sodium channels investigated with μ-conotoxins and the recently discovered μO§-conotoxin GVIIJ. J Neurophysiol, 2015; 113: 2289-2301.[DOI]
[202] Fainzilber M, Nakamura T, Gaathon A et al. A new cysteine framework in sodium channel blocking conotoxins. Biochemistry, 1995; 34: 8649-8656.[DOI]
[203] Hasson A, Fainzilber M, Zlotkin E et al. Electrophysiological characterization of a novel conotoxin that blocks molluscan sodium channels. Eur J Neurosci, 1995; 7: 815-818.[DOI]
[204] Liu J, Wu Q, Pi C et al. Isolation and characterization of a T-superfamily conotoxin from Conus litteratus with targeting tetrodotoxin-sensitive sodium channels. Peptides, 2007; 28: 2313-2319.[DOI]
[205] Yang M, Zhou M. μ-conotoxin TsIIIA, a peptide inhibitor of human voltage-gated sodium channel hNa(v)1.8. Toxicon, 2020; 186: 29-34.[DOI]
[206] Yang M, Zhao S, Min X et al. A novel μ-conotoxin from worm-hunting Conus tessulatus that selectively inhibit rat TTX-resistant sodium currents. Toxicon, 2017; 130: 11-18.[DOI]
[207] McMahon KL, Tran HNT, Deuis JR et al. µ-Conotoxins Targeting the Human Voltage-Gated Sodium Channel Subtype Na(V)1.7. Toxins, 2022; 14.[DOI]
[208] McIntosh JM, Hasson A, Spira ME et al. A new family of conotoxins that blocks voltage-gated sodium channels. J Biol Chem, 1995; 270: 16796-16802.[DOI]
[209] Terlau H, Stocker M, Shon KJ et al. MicroO-conotoxin MrVIA inhibits mammalian sodium channels, but not through site I. J Neurophysiol, 1996; 76: 1423-1429.[DOI]
[210] Daly NL, Ekberg JA, Thomas L et al. Structures of muO-conotoxins from Conus marmoreus. I nhibitors of tetrodotoxin (TTX)-sensitive and TTX-resistant sodium channels in mammalian sensory neurons. J Biol Chem, 2004; 279: 25774-25782.[DOI]
[211] de Araujo AD, Callaghan B, Nevin ST et al. Total synthesis of the analgesic conotoxin MrVIB through selenocysteine-assisted folding. Angew Chem Int Ed Engl, 2011; 50: 6527-6529.[DOI]
[212] Vetter I, Dekan Z, Knapp O et al. Isolation, characterization and total regioselective synthesis of the novel μO-conotoxin MfVIA from Conus magnificus that targets voltage-gated sodium channels. Biochem Pharmacol, 2012; 84: 540-548.[DOI]
[213] Gajewiak J, Azam L, Imperial J et al. A disulfide tether stabilizes the block of sodium channels by the conotoxin μO§-GVIIJ. PNAS, 2014; 111: 2758-2763.[DOI]
[214] Wang D, Himaya SWA, Giacomotto J et al. Characterisation of d-Conotoxin TxVIA as a Mammalian T-Type Calcium Channel Modulator. Mar Drugs, 2020; 18.[DOI]
[215] Hasson A, Fainzilber M, Gordon D et al. Alteration of sodium currents by new peptide toxins from the venom of a molluscivorous Conus snail. Eur J Neurosci, 1993; 5: 56-64.[DOI]
[216] Jin AH, Israel MR, Inserra MC et al. δ-Conotoxin SuVIA suggests an evolutionary link between ancestral predator defence and the origin of fish-hunting behaviour in carnivorous cone snails. Proc Biol Sci, 2015; 282.[DOI]
[217] Aman JW, Imperial JS, Ueberheide B et al. Insights into the origins of fish hunting in venomous cone snails from studies of Conus tessulatus. PNAS, 2015; 112: 5087-5092.[DOI]
[218] Bulaj G, DeLaCruz R, Azimi-Zonooz A et al. Delta-conotoxin structure/function through a cladistic analysis. Biochemistry, 2001; 40: 13201-13208.[DOI]
[219] Leipold E, Hansel A, Olivera BM et al. Molecular interaction of delta-conotoxins with voltage-gated sodium channels. FEBS Lett, 2005; 579: 3881-3884.[DOI]
[220] Green BR, Gajewiak J, Chhabra S et al. Structural Basis for the Inhibition of Voltage-gated Sodium Channels by Conotoxin μO§-GVIIJ. J Biol Chem, 2016; 291: 7205-7220.[DOI]
[221] Zhang MM, Gajewiak J, Azam L et al. Probing the Redox States of Sodium Channel Cysteines at the Binding Site of μO§-Conotoxin GVIIJ. Biochemistry, 2015; 54: 3911-3920.[DOI]
[222] Hillyard DR, Olivera BM, Woodward S et al. A molluscivorous Conus toxin: conserved frameworks in conotoxins. Biochemistry, 1989; 28: 358-361.[DOI]
[223] Fainzilber M, Gordon D, Hasson A et al. Mollusc-specific toxins from the venom of Conus textile neovicarius. Eur J Biochem, 1991; 202: 589-595.[DOI]
[224] Nakamura T, Yu Z, Fainzilber M et al. Mass spectrometric-based revision of the structure of a cysteine-rich peptide toxin with gamma-carboxyglutamic acid, TxVIIA, from the sea snail, Conus textile. Protein Sci, 1996; 5: 524-530.[DOI]
[225] Fainzilber M, Lodder JC, Kits KS et al. A new conotoxin affecting sodium current inactivation interacts with the delta-conotoxin receptor site. J Biol Chem, 1995; 270: 1123-1129.[DOI]
[226] Peigneur S, Paolini-Bertrand M, Gaertner H et al. δ-Conotoxins synthesized using an acid-cleavable solubility tag approach reveal key structural determinants for NaV subtype selectivity. J Biol Chem, 2014; 289: 35341-35350.[DOI]
[227] Terlau H, Shon KJ, Grilley M et al. Strategy for rapid immobilization of prey by a fish-hunting marine snail. Nature, 1996; 381: 148-151.[DOI]
[228] Teichert RW, Jacobsen R, Terlau H et al. Discovery and characterization of the short kappaA-conotoxins: a novel subfamily of excitatory conotoxins. Toxicon, 2007; 49: 318-328.[DOI]
[229] Santos AD, McIntosh JM, Hillyard DR et al. The A-superfamily of conotoxins: structural and functional divergence. J Biol Chem, 2004; 279: 17596-17606.[DOI]
[230] Cordeiro S, Finol-Urdaneta RK, Köpfer D et al. Conotoxin κM-RIIIJ, a tool targeting asymmetric heteromeric K(v)1 channels. PNAS, 2019; 116: 1059-1064.[DOI]
[231] Al-Sabi A, Lennartz D, Ferber M et al. KappaM-conotoxin RIIIK, structural and functional novelty in a K+ channel antagonist. Biochemistry, 2004; 43: 8625-8635.[DOI]
[232] Fan C-X, Chen X-K, Zhang C et al. A Novel Conotoxin from Conus betulinus, κ-BtX, Unique in Cysteine Pattern and in Function as a Specific BK Channel Modulator*. J Biol Chem, 2003; 278: 12624-12633.[DOI]
[233] Imperial JS, Bansal PS, Alewood PF et al. A novel conotoxin inhibitor of Kv1.6 channel and nAChR subtypes defines a new superfamily of conotoxins. Biochemistry, 2006; 45: 8331-8340.[DOI]
[234] Martínez-Hernández L, López-Vera E, Aguilar MB et al. κO-SrVIA Conopeptide, a Novel Inhibitor Peptide for Two Members of the Human EAG Potassium Channel Family. Int J Mol Sci, 2023; 24: 11513.[DOI]
[235] Kauferstein S, Huys I, Lamthanh H et al. A novel conotoxin inhibiting vertebrate voltage-sensitive potassium channels. Toxicon, 2003; 42: 43-52.[DOI]
[236] McCoy MT, Jayanthi S, Cadet JL. Potassium Channels and Their Potential Roles in Substance Use Disorders. Int J Mol Sci, 2021; 22: 1249.[DOI]
[237] Martínez-Hernández L, López-Vera E, Aguilar MB. Peptide Toxins from Marine Conus Snails with Activity on Potassium Channels and/or Currents. Toxins, 2024; 16: 504.[DOI]
[238] Jiang X, Liu J. Handbook of experimental pharmacology. , 2013.
[239] Burg S, Attali B. Targeting of potassium channels in cardiac arrhythmias. Trends Pharmacol Sci, 2021; 42: 491-506.[DOI]
[240] Ferber M, Sporning A, Jeserich G et al. A novel Conus peptide ligand for K+ channels. J Biol Chem, 2003; 278: 2177-2183.[DOI]
[241] Al-Sabi A, Lennartz D, Ferber M et al. κM-conotoxin RIIIK, structural and functional novelty in a K+ channel antagonist. Biochemistry, 2004; 43: 8625-8635.[DOI]
[242] Ferber M, Al-Sabi A, Stocker M et al. Identification of a mammalian target of κM-conotoxin RIIIK. Toxicon, 2004; 43: 915-921.[DOI]
[243] Chen P, Dendorfer A, Finol-Urdaneta RK et al. Biochemical characterization of κM-RIIIJ, a Kv1. 2 channel blocker: Evaluation of cardioprotective effects of κM-conotoxins. J Biol Chem, 2010; 285: 14882-14889.[DOI]
[244] Cordeiro S, Finol-Urdaneta RK, Köpfer D et al. Conotoxin κM-RIIIJ, a tool targeting asymmetric heteromeric Kv1 channels. PNAS, 2019; 116: 1059-1064.[DOI]
[245] Judge SI, Bever Jr CT. Potassium channel blockers in multiple sclerosis: neuronal Kv channels and effects of symptomatic treatment. Pharmacol Therapeut, 2006; 111: 224-259.[DOI]
[246] Ramiro IBL, Bjørn-Yoshimoto WE, Imperial JS et al. Somatostatin venom analogs evolved by fish-hunting cone snails: From prey capture behavior to identifying drug leads. Sci Adv, 2022; 8: eabk1410.[DOI]
[247] Heading CE. Ziconotide (Elan Pharmaceuticals). IDrugs, 2001; 4: 339-350.
[248] Miljanich GP. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr Med Chem, 2004; 11: 3029-3040.[DOI]
[249] Gao S, Yao X, Yan N. Structure of human Ca(v)2.2 channel blocked by the painkiller ziconotide. Nature, 2021; 596: 143-147.[DOI]
[250] Deer TR, Pope JE, Hayek SM et al. The Polyanalgesic Consensus Conference (PACC): Recommendations on Intrathecal Drug Infusion Systems Best Practices and Guidelines. Neuromodulation, 2017; 20: 96-132.[DOI]
[251] Lambe T, Duarte R, Eldabe R et al. Ziconotide for the Management of Cancer Pain: A Budget Impact Analysis. Neuromodulation, 2023; 26: 1226-1232.[DOI]
[252] Otvos L, Jr., Wade JD. Big peptide drugs in a small molecule world. Front Chem, 2023; 11: 1302169.[DOI]
[253] Korte A. 2022 Golden Goose Award honors serendipitous science. Science, 2022; 377: 1500-1501.[DOI]
[254] Lee HK, Zhang L, Smith MD et al. A marine analgesic peptide, Contulakin-G, and neurotensin are distinct agonists for neurotensin receptors: uncovering structural determinants of desensitization properties. Front Pharmacol, 2015; 6: 11.[DOI]
[255] Sang CN, Barnabe KJ, Kern SE. Phase IA Clinical Trial Evaluating the Tolerability, Pharmacokinetics, and Analgesic Efficacy of an Intrathecally Administered Neurotensin A Analogue in Central Neuropathic Pain Following Spinal Cord Injury. Clin Pharmacol Drug Dev, 2016; 5: 250-258.[DOI]
[256] Allen JW, Hofer K, McCumber D et al. An assessment of the antinociceptive efficacy of intrathecal and epidural contulakin-G in rats and dogs. Anesth Analg, 2007; 104: 1505-1513, table of contents.[DOI]
[257] Kern SE, Allen J, Wagstaff J et al. The pharmacokinetics of the conopeptide contulakin-G (CGX-1160) after intrathecal administration: an analysis of data from studies in beagles. Anesth Analg, 2007; 104: 1514-1520.[DOI]
[258] Martin L, Ibrahim M, Gomez K et al. Conotoxin contulakin-G engages a neurotensin receptor 2/R-type calcium channel (Cav2.3) pathway to mediate spinal antinociception. Pain, 2022; 163: 1751-1762.[DOI]
[259] Martin LF, Almuslim M, Ismail KA et al. The conotoxin Contulakin-G reverses hypersensitivity observed in rodent models of cancer-induced bone pain without inducing tolerance or motor disturbance. Pain, 2024.[DOI]
[260] Sandall DW, Satkunanathan N, Keays DA et al. A novel alpha-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry, 2003; 42: 6904-6911.[DOI]
[261] Romero HK, Christensen SB, Di Cesare Mannelli L et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. PNAS, 2017; 114: E1825-e1832.[DOI]
[262] Christensen SB, Hone AJ, Roux I et al. RgIA4 Potently Blocks Mouse α9α10 nAChRs and Provides Long Lasting Protection against Oxaliplatin-Induced Cold Allodynia. Front Cell Neurosci, 2017; 11.[DOI]
[263] Huynh PN, Giuvelis D, Christensen S et al. RgIA4 Accelerates Recovery from Paclitaxel-Induced Neuropathic Pain in Rats. Mar Drugs, 2019; 18.[DOI]
[264] Huynh PN, Christensen SB, McIntosh JM. RgIA4 Prevention of Acute Oxaliplatin-Induced Cold Allodynia Requires α9-Containing Nicotinic Acetylcholine Receptors and CD3+ T-Cells. Cells, 2022; 11: 3561.[DOI]
[265] Zheng N, Christensen SB, Blakely A et al. Development of conformationally constrained α-RgIA analogues as stable peptide antagonists of human α9α10 nicotinic acetylcholine receptors. J Med Chem, 2020; 63: 8380-8387.[DOI]
[266] Gajewiak J, Christensen SB, Dowell C et al. Selective penicillamine substitution enables development of a potent analgesic peptide that acts through a non-opioid-based mechanism. J Med Chem, 2021; 64: 9271-9278.[DOI]
[267] Xiong X, Menting JG, Disotuar MM et al. A structurally minimized yet fully active insulin based on cone-snail venom insulin principles. Nat Struct Mol Biol, 2020; 27: 615-624.[DOI]
[268] Terlau H, Boccaccio A, Olivera BM et al. The block of Shaker K+ channels by kappa-conotoxin PVIIA is state dependent. J Gen Physiol, 1999; 114: 125-140.[DOI]
[269] Zhang SJ, Yang XM, Liu GS et al. CGX-1051, a peptide from Conus snail venom, attenuates infarction in rabbit hearts when administered at reperfusion. J Cardiovasc Pharmacol, 2003; 42: 764-771.[DOI]
[270] Lubbers NL, Campbell TJ, Polakowski JS et al. Postischemic administration of CGX-1051, a peptide from cone snail venom, reduces infarct size in both rat and dog models of myocardial ischemia and reperfusion. J Cardiovasc Pharmacol, 2005; 46: 141-146.[DOI]
[271] Rashid MH, Mahdavi S, Kuyucak S. Computational Studies of Marine Toxins Targeting Ion Channels. Mar Drugs, 2013; 11: 848-869.[DOI]
[272] Luo S, Zhangsun D, Harvey PJ et al. Cloning, synthesis, and characterization of αO-conotoxin GeXIVA, a potent α9α10 nicotinic acetylcholine receptor antagonist. PNAS, 2015; 112: E4026-4035.[DOI]
[273] Wang H, Li X, Zhangsun D et al. The α9α10 Nicotinic Acetylcholine Receptor Antagonist αO-Conotoxin GeXIVA[1,2] Alleviates and Reverses Chemotherapy-Induced Neuropathic Pain. Mar Drugs, 2019; 17.[DOI]
[274] Wang H, Li X, Qiao Y et al. αO-Conotoxin GeXIVA[1,2] Reduced Neuropathic Pain and Changed Gene Expression in Chronic Oxaliplatin-Induced Neuropathy Mice Model. Mar Drugs, 2024; 22.[DOI]
[275] Liu Y, Qian J, Sun Z et al. Cervical Cancer Correlates with the Differential Expression of Nicotinic Acetylcholine Receptors and Reveals Therapeutic Targets. Mar Drugs, 2019; 17.[DOI]
[276] Ju S, Zhang Y, Guo X et al. Anti-Ovarian Cancer Conotoxins Identified from Conus Venom. Molecules, 2022; 27.[DOI]
[277] Sun Z, Bao J, Zhangsun M et al. αO-Conotoxin GeXIVA Inhibits the Growth of Breast Cancer Cells via Interaction with α9 Nicotine Acetylcholine Receptors. Mar Drugs, 2020; 18.[DOI]
[278] Guo X, He L, Xu W et al. αO-Conotoxin GeXIVA[1,2] Suppresses In Vivo Tumor Growth of Triple-Negative Breast Cancer by Inhibiting AKT-mTOR, STAT3 and NF-κB Signaling Mediated Proliferation and Inducing Apoptosis. Mar Drugs, 2024; 22.[DOI]
[279] Robinson SD, Undheim EAB, Ueberheide B et al. Venom peptides as therapeutics: advances, challenges and the future of venom-peptide discovery. Expert Rev Proteomics, 2017; 14: 931-939.[DOI]
[280] Li Q, Watkins M, Robinson SD et al. Discovery of Novel Conotoxin Candidates Using Machine Learning. Toxins, 2018; 10.[DOI]
[281] Monroe LK, Truong DP, Miner JC et al. Conotoxin Prediction: New Features to Increase Prediction Accuracy. Toxins, 2023; 15: 641.[DOI]
[282] Truong DP, Monroe LK, Williams RF et al. Machine Learning Framework for Conotoxin Class and Molecular Target Prediction. Toxins, 2024; 16: 475.[DOI]
[283] Guo M, Li Z, Deng X et al. ConoDL: a deep learning framework for rapid generation and prediction of conotoxins. J Comput Aided Mol Des, 2024; 39: 4.[DOI]
[284] Wu X, Hone AJ, Huang YH et al. Computational Design of α-Conotoxins to Target Specific Nicotinic Acetylcholine Receptor Subtypes. Chemistry, 2024; 30: e202302909.[DOI]
[285] Hone AJ, McIntosh JM. Nicotinic acetylcholine receptors: Therapeutic targets for novel ligands to treat pain and inflammation. Pharmacol Res, 2023; 190: 106715.[DOI]
[286] Australian Government Federal Register of Legislation Defence and Strategic Goods List 2024. Accessed on 14 January 2024. Available at: [Web]
[287] US Centers for Disease Control and Prevention Select Agents and Toxins List. Accessed on 14 January 2024. Available at: [Web]
[288] The Australia Group. Accessed on 14 January 2024. Available at: [Web]
[289] Johnson KA. Preparation of peptide and protein powders for inhalation. Adv Drug Deliv Rev, 1997; 26: 3-15.[DOI]
[290] Gao B, Peng C, Yang J et al. Cone Snails: A Big Store of Conotoxins for Novel Drug Discovery. Toxins, 2017; 9.[DOI]
[291] Peters H, O'Leary BC, Hawkins JP et al. Conus: First Comprehensive Conservation Red List Assessment of a Marine Gastropod Mollusc Genus. PLoS One, 2013; 8: e83353.[DOI]
[292] Dutertre S, Lewis R. Cone snail Biology, Bioprospecting and Conservation. In: Hämäläinen EM, Järvinen S (ed). Snails: Biology, Ecology, and Conservation. Nova Science Publishers, Hauppauge, USA. , 2013: 85-104.
[293] Clark C, Olivera BM, Cruz LJ. A toxin from the venom of the marine snail Conus geographus which acts on the vertebrate central nervous system. Toxicon, 1981; 19: 691-699.[DOI]
[294] McIntosh M, Cruz LJ, Hunkapiller MW et al. Isolation and structure of a peptide toxin from the marine snail Conus magus. Arch Biochem Biophys, 1982; 218: 329-334.[DOI]
[295] Cruz LJ, Gray WR, Olivera BM. Purification and properties of a myotoxin from Conus geographus venom. Arch Biochem Biophys, 1978; 190: 539-548.[DOI]
[296] Milisen JW. Conopeptide production through biosustainable snail farming University of Hawai'i at Manoa; 2012.
[297] Petuch E, Berschauer D. A New Species of Jaspidiconus (Gastropoda: Conidae) from Aruba. 2022; 54: 169-171.[DOI]
[298] Slapcinsky J, Murphy AE. Overlooked predatory snails from Papua New Guinea: nine new Torresiropa Solem, 1959 (Gastropoda: Stylommatophora: Rhytididae). Archiv für Molluskenkunde Int J Malacol, 2023: 135-152.[DOI]
[299] Rogalski A, Himaya SWA, Lewis RJ. Coordinated adaptations define the ontogenetic shift from worm- to fish-hunting in a venomous cone snail. Nat Commun, 2023; 14: 3287.[DOI]
[300] Ho TN, Tran TH, Le HS et al. Advances in the synthesis and engineering of conotoxins. Eur J Med Chem, 2025; 282: 117038.[DOI]
[301] Han TS, Teichert RW, Olivera BM et al. Conus venoms - a rich source of peptide-based therapeutics. Curr Pharm Des, 2008; 14: 2462-2479.[DOI]
[302] Gao F, Chen D, Ma X et al. Alpha6-containing nicotinic acetylcholine receptor is a highly sensitive target of alcohol. Neuropharmacology, 2019; 149: 45-54.[DOI]


