Oncolytic Viruses: From Basics to Challenges to Innovation
Ziyad S. Haidar1,2,3,4,5*
1BioMAT’X I+D+i (HAiDAR R&D&I) LAB, Universidad de los Andes, Santiago, Chile
2Programa de Doctorado en BioMedicina, Facultad de Medicina, Universidad de los Andes, Santiago, Chile
3Programa de Doctorado en Ciencias Odontológicas, Facultad de Odontología, Universidad de los Andes, Santiago, Chile
4Centro de Investigación e Innovación Biomédica (CiiB), Universidad de los Andes, Santiago, Chile
5Facultad de Odontología, Universidad de los Andes, Santiago, Chile
*Correspondence to: Ziyad S. Haidar, PhD, Professor, BioMAT’ X I+D+i (HAiDAR R&D&I) LAB, University of the Andes, Santiago, 8620000, Chile; Email: zhaidar@uandes.cl
DOI: 10.53964/id.2024027
Abstract
The landscape of cancer therapeutics has undergone a transformative shift from broad approaches, such as radiation and chemotherapy, to more precise strategies, encompassing small molecule kinase inhibitors and monoclonal antibodies targeting immune checkpoint molecules. Oncolytic viruses (OVs) have recently emerged as a promising and viable option for cancer immunotherapy, particularly for “cold” tumors entrenched in an immunologically-suppressive tumor microenvironment. Genetic attenuation refines the characteristics of OVs, replacing specific genes to enhance conditional viral replication within tumor cells. Despite their potential, the therapeutic use of OVs faces challenges such as pre-mature elimination by the immune system of the host alongside the dense stromal barriers. In this review article, the strategic encapsulation of genetically-engineered OVs within intra-lesional/-tumoral carriers/ vehicles such as mesenchymal stem cells are explored. To further enhance vector delivery strategies and precise targeting, protecting/shielding OVs from host immune responses, via utilizing release-controlled nanoparticles and employing nanofilaments to optimize vector propagation, for example, are discussed. Such innovative approaches not only mitigate challenges related to pre-mature viral clearance and stromal barriers yet also facilitates a localized, controlled, and sustained release of OVs, thereby optimizing their therapeutic bio-safety and -impact. Ongoing clinical investigations are exploring the synergies between OVs and carriers/delivery systems, combined with various anti-cancer therapeutics as radiotherapy, chemotherapy, immune checkpoint inhibitors, and monoclonal antibodies. These studies hold promise for revolutionizing cancer treatment, ensuring both safety and efficacy, aiming to advance bio-pharmaceutical delivery systems from bench-top to bed-side for enhanced therapeutic results, improved patient survival, and enhanced quality of life or QoL.
Keywords: oncolytic virus, cancer, tumor, stroma, drug delivery, immunotherapy, controlled release
1 INTRODUCTION
Cancer remains a predominant cause of mortality, posing a significant threat to human health, and global predictions by the World Health Organization indicate a continual rise in cancer incidence and mortality over the next two decades[1]. Despite advancements in traditional therapeutic approaches, including surgery, chemotherapy, radiation therapy, and emerging immunotherapy, their efficacy is not optimal. Consequently, there exists a pressing need for innovative anti-tumoral strategies. Vaccines have emerged as pivotal advancements in immunology and healthcare, leveraging the inoculation of attenuated or noninfectious components of infectious agents to trigger an immune response. By programming the immune system to recognize and target foreign pathogens, vaccines confer protective immunity, enabling a swift and efficient immune response upon subsequent exposure to the authentic virus, thereby preventing severe infections[2]. Notably, vaccine development has played a crucial role in significantly reducing mortality associated with diseases like smallpox, yellow fever[3], measles, mumps, rubella, and varicella[4]. Certain viruses exhibit a remarkable ability to target tumor cells, giving rise to oncolytic viruses (OVs). OVs, often live and infectious viruses, have garnered interest in cancer therapeutics due to their capacity for inducing selective cell death and fostering specific anti-tumor immunity. This review comprehensively summarizes the diverse landscape of OVs documented in the literature, addresses the challenges within oncolytic viral therapy, and proposes strategies for modifying and integrating OVs with traditional cancer therapies to enhance the overall success of adjuvant treatments and improve the quality of life of patients.
2 CHARACTERISTIC(S) OF OVS
Briefly, OVs represent a category of cancer therapies characterized by the deliberate selection of viruses for their oncolytic capabilities. Typically, these viruses undergo attenuation through modifications in their genome, resulting in reduced cytotoxicity towards non-cancerous cells and conditional replication within cancer cells. Alternatively, OVs may be selected through multiple passages in tumor tissues, where key viral genes essential for virulence are replaced with genes encoding proteins designed to specifically target tumor cells. This strategic approach ensures that the virus avoids targeting nonmalignant tissues and confines its replication solely to within tumor cells[5,6]. While the use of engineered viruses in virotherapy raises concerns about potential insertional mutagenesis, wherein the viral genome integrates into the host’s genome[7], OVs undergo extensive preclinical studies to evaluate their efficacy and safety before human application. In 2008, Duerner et al.[8] study demonstrated the conditionally replication-competent murine leukemia virus to serve as an example of this pre-clinical assessment. Despite the low probability of genomic integration and adverse effects in clinical outcomes, the imperative need for robust, long-term safety data arises as more therapeutic applications incorporate these engineered viruses in both oncology and non-oncology settings[7]. The efficacy of selectively eliminating cancer cells often hinges on factors such as the viral strain, cancer type, tumor microenvironment (TME), and the immune system of the host. OVs are intended to preferentially or selectively target cancer cells by leveraging distinctive extracellular surface markers present on these cells, facilitating their entry. Notably, frequently overexpressed surface markers on tumor cells such as CD46, CD155, and integrin α2β1 act as receptors for measles virus, poliovirus, and echovirus, respectively[9,10]. Cancer cells often exhibit specific mutations, such as aberrations in BCL-2, EGFR, PTEN, RAS, RB1, TP53, and WNT, leading to uncontrolled cell proliferation. Interestingly, these mutations may render tumor cells susceptible to viral infection, resulting in subsequent cytotoxic elimination[11-13]. In contrast to normal cells that induce interferon (IFN) expression in response to viral infection, cancer cells, due to their inability to trigger type 1 IFN signaling, allow OVs to replicate freely within them. This, in turn, induces oncolysis and the release of viral progeny to perpetuate the infection cycle. Additionally, OVs can be engineered to express immunostimulatory cytokines / chemokines, such as tumor necrotic factor (TNF), IFN α, and granulocyte-macrophage colony-stimulating factor (GM-CSF), enhancing their ability to provoke a robust host immune response[3,4]. OV treatment initiates with the introduction of the virus, triggering subsequent viral replication that induces extensive damage within cancer cells, ultimately compromising their integrity and leading to oncolysis[11]. Beyond oncolysis, OV replication plays a pivotal role in fostering robust anti-tumor immunity by inducing immunogenic cell death (ICD). This process releases various components, including tumor antigens (TA), damage-associated molecular patterns (DAMPs), OV-derived pathogen-associated molecular patterns (PAMPs), and inflammatory cytokines, effectively activating and recruiting both innate and adaptive immune cells[14,15]. PAMPs serve to alert the immune system to the presence of pathogens[16], while DAMPs contribute to signaling tissue trauma, binding to corresponding receptors on dendritic cells and inducing T-cell activation. This dynamic interplay significantly influences the immune balance within the TME[17,18]. Additionally, certain OVs can stimulate the anti-tumor response without relying on viral replication-mediated oncolysis. When OVs bind to tumor cells, they activate an antiviral immune response, where PAMPs and DAMPs prompt cytokine secretion, recruiting immune cells to the site. This alternative pathway also effectively promotes an anti-tumor immune response (Figure 1)[19,20]. Therefore, OVs emerge as a viable strategy to target notoriously non-immunogenic “cold” tumors, which reside in a TME suppressing immune responses and favoring T cell invasion. This approach proves effective in stimulating both the innate and adaptive immune systems, especially crucial for cold tumors that often exhibit non-responsiveness to currently available immune checkpoint inhibitors. The imperative for enhancing the efficacy of cancer immunotherapy in these challenging cases is evident[21,22].
|
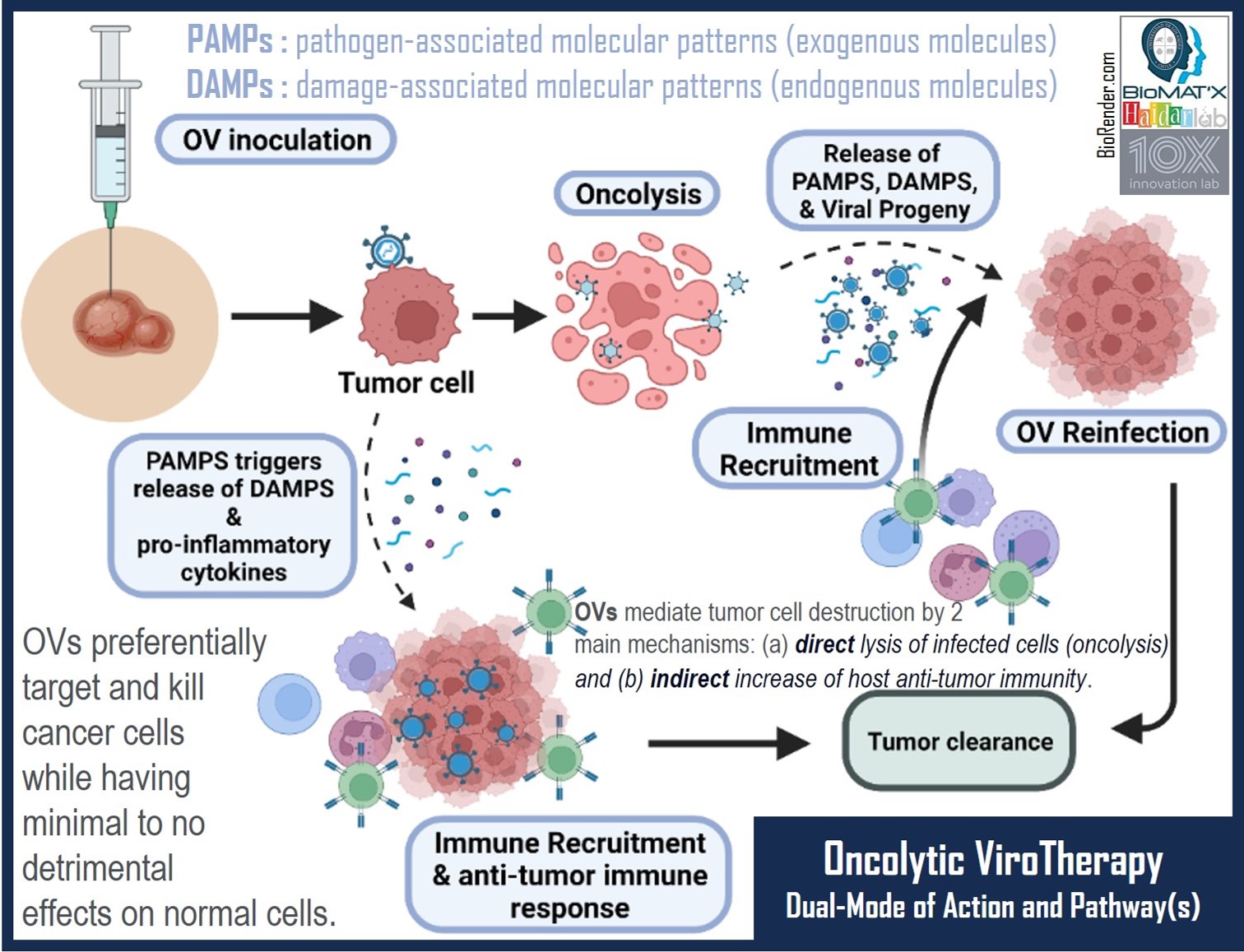
Figure 1. Pathway of/for the Dual-Mode of Action of Oncolytic viroTherapy: PAMPs and DAMPs. The process begins with the inoculation of the OVs into the tumor. OVs selectively bind to unique extracellular surface markers expressed exclusively on tumor cells, facilitating cellular entry. Once inside, OVs hijack the host machinery, rapidly replicate, and induce oncolysis, releasing viral progeny, pathogen-associated molecular patterns (PAMPs that alert the immune system to the presence of pathogens), damage-associated molecular patterns (DAMPs that signal tissue trauma and activate dendritic cells), chemokines, and cytokines. The released viral progeny continues and perpetuates the oncolytic cycle by binding to neighboring / adjacent tumor cells, while the other accompanying factors work synergistically to recruit various types of immune cells (e.g., CD4+ T cells, CD8+ T cells, and NK cells) to the tumor (site), allowing for tumor infiltration and enhanced eradication of malignant cells. Alternatively, and interestingly, some OVs, rather than inducing oncolysis, trigger the secretion of DAMPs and pro-inflammatory cytokines, effectively recruiting immune cells to target the tumor cells. This orchestrated immune response, bolstered by the release of PAMPs and DAMPs, amplifies the therapeutic impact of oncolytic virotherapy. Illustration created using BioRender.com.
3 STRAINS OF OVS
The first OV to be approved by a regulatory agency was a genetically modified adenovirus named H101 by Shanghai Sunway Biotech. Different types of oncolytic viruses - adenoviruses, alphaviruses, herpes simplex viruses, Newcastle disease viruses, rhabdoviruses, Coxsackie viruses, and vaccinia viruses - have been applied as either naturally occurring or engineered vectors. Exploration of various OVs with anti-tumor properties encompasses both DNA and RNA viruses, as detailed in Table 1. It is crucial to distinguish OVs from other genetically modified viruses used for treatment, wherein the defining feature is the ability to replicate and induce cell lysis. Genetically modified viruses, like certain adenoviral agents, function as viral vectors, delivering genes (often tumor antigens) but are replication-defective (RD), ensuring treatment safety[23]. This characteristic is commonly employed in vaccine development, exemplified by RD-recombinant chimpanzee adenovirus type 3-vectored ebolavirus vaccine (cAd3-EBO) and other studies using adenoviral vectors in vaccine platforms[24]. DNA viruses, benefiting from higher genome stability due to high-fidelity DNA polymerases, possess larger genomes allowing for greater transgene insertions without compromising infection and replication capacity, albeit at the cost of impeding replication kinetics[25-29]. While DNA viruses can elicit strong antiviral responses and anti-tumor immunity, the presence of high neutralizing antibodies may limit viral replication, although some OVs demonstrate efficient replication even in the presence of nAbs targeting the backbone virus[28]. Conversely, RNA viruses like NDV, poliovirus, and reovirus exhibit limited genomic packaging capacity but heightened immunogenicity. RNA viruses replicate in the cytoplasm, demonstrating rapid proliferation with high mutation rates, facilitating rapid evolution toward an oncolytic phenotype but also posing a risk of divergence from this desired characteristic[25,27]. Genetic instability in RNA viruses could potentially serve as an advantage in “personalized” targeted therapy, allowing for multiple optimized virus variants to promote tumor clearance in the presence of anti-viral immunity[30]. The use of RNA viruses, therefore, requires careful consideration and judicious design in the construction of OVs and study protocols. As mentioned earlier, prominent or common examples of oncolytic DNA viruses encompass, including vaccinia virus (VV), adenoviruses, and herpes simplex virus (HSV). VV, a double-stranded DNA (dsDNA) virus, infects and replicates within the cytoplasm of mammalian cells[31]. Various vaccinia virus agents have undergone scrutiny, with Pexa-Vec standing out. Pexa-Vec is an oncolytic VV characterized by an inactivated thymidine kinase (TK) gene, replaced with a transgene expressing human GM-CSF and β-galactosidase[32]. Its efficacy has been assessed in treating hepatocellular carcinoma (HCC) and colorectal cancer[31,33,34]. Additionally, clinical investigations involve GL-ONC1 (VV incorporating Ruc-GFP, β-glucuronidase, and β-galactosidase transgene insertions), vvDD (VV with deletion of the vaccinia growth factor and TK genes), and TBio-6517 (VV expressing Flt3 ligand, the cytokine IL-12, and an antibody targeting CTLA4)[35-37]. Adenovirus, a dsDNA virus, saw its inaugural human testing with Onyx-015 (lontucirev). This recombinant adenovirus exhibits viral attenuation and conditional replication achieved through the deletion of the E1B locus, responsible for encoding the 55kDa E1B protein[38]. Despite promising results in the interim efficacy and safety report during phase III trials for head and neck cancer in China, Onyx-015 was discontinued[39]. Second-generation adenoviruses, exemplified by DNX-2401 (tasadenoturev), have shown efficacy against glioblastomas[40]. With a 24-base pair deletion in the E1A gene, DNX-2401 selectively targets cancer cells featuring abnormalities in the retinoblastoma (Rb) pathway[41]. Notably, it received fast-track orphan drug designation from the U.S. Food and Drug Administration (FDA) in 2014 for malignant glioma treatment and is currently being explored in combination with immune checkpoint inhibitors. Other oncolytic adenoviruses undergoing clinical investigation include enadenotucirev (chimeric Ad11p / Ad3 oncolytic adenovirus with a 25bp deletion of E4 and 2,444bp deletion in E3ORF), LOAd703 (serotype 5 adenovirus with serotype 35 fiber and knob, encoding trimerized membrane-bound CD40L and 4-1BBL), and ONCOS-102 (modified serotype 5 adenovirus with a serotype 3 knob, insertion of the GM-CSF transgene, and a 24bp deletion of the Rb binding site of the E1A gene)[42-45]. HSV, encompassing both HSV-1 and HSV-2, is a dsDNA virus with a natural propensity to infect humans[46,47]. HSV has found application in oncolytic therapy targeting melanomas, gliomas, and colorectal cancer[48,49]. The mutant strain HSV1716, devoid of the ICP34.5 neurovirulence gene, exhibits selective targeting and replication in human glioblastoma cells[50]. Another mutant HSV, NV1020, involves deletions in a 15-kb region at the UL/S junction, including the UL56 gene, and further attenuation through a 700-bp deletion encompassing the TK gene and the UL24 promoter[51,52]. Reintroduction of the viral HSV-1 TK gene allows for the control of NV1020 infection using TK-converted prodrugs like acyclovir. Weekly hepatic arterial infusion of NV1020 demonstrated the stabilization of liver metastasis in 50% of patients with heavily treated colorectal cancer at the optimal biological dose of 1×108 plaque-forming units (PFU)[49]. Additional HSV-based OVs undergoing clinical trials include G207 (an HSV-1 strain with deletion of the neurovirulent γ134.5 gene and insertion of β-galactosidase to inactivate the UL39 gene), ONCR-177 (an HSV-1 agent with a mutant UL37 gene, tissue-specific miRNA attenuation, and insertion of five transgenes for IL-12, FLT3LG, CCL2, and antagonists against programmed death protein 1 (PD-1) and CTLA-4), OH2 (genetically modified HSV-2 expressing GM-CSF), and RP1 (an HSV-1 agent expressing GM-CSF)[53-57]. NDV is an avian host-targeting single-stranded RNA (ssRNA) virus that naturally infects poultry[58,59]. Among the well-studied strains of NDV, MTH-68/H has been employed in the treatment of epithelial tumors and high-grade glioma[48,60,61]. Another NDV variant, LaSota, exhibits a lentogenic strain of lower pathogenicity. In vitro studies using Human Papilloma Virus (HPV) E6/E7 expressing TC-1 cells, a cervical cancer model, revealed suppressed tumor growth through oncolytic virus-induced apoptosis[61]. Poliovirus, a naturally neuron-targeting ssRNA virus, serves as an effective vehicle for glioma-targeted oncolytic therapies. Restricted to human and old-world primates, poliovirus enters host cells through viral binding with the poliovirus receptor Nectin-like molecule 5 (Necl-5) or CD15[62,63]. The recombinant virus PVSRIPO, a chimera created from non-pathogenic strains of rhinovirus and type 1 poliovirus vaccine, has been studied in malignant glioma and melanoma[64,65]. Replacement of the poliovirus internal ribosomal entry site (IRES) with that of rhinovirus attenuates neurovirulence[66] and facilitates conditional replication in tumor cells, specifically binding to CD155, which is highly upregulated in many cancer types[63,67]. Respiratory enteric orphan virus (reovirus)[68], a non-enveloped, dsDNA virus, exhibits preferential replication within cancer cells expressing a constitutively activated Ras pathway[69], without affecting nonmalignant cells lacking Ras activation[70]. Pelareorep, a truncated form of reovirus, obtained orphan drug status in 2015 for the treatment of malignant gliomas, ovarian cancer, and pancreatic cancer, considered Ras-activated tumors[71]. Subsequently, reovirus has been applied to treat melanomas, breast cancer, and head and neck squamous cell carcinoma[72-74].
Table 1. DNA Versus RNA Viral Characteristics with a Brief Overview of Viruses Developed as Oncolytic Vectors
|
Genome |
|
DNA Viruses |
RNA Viruses |
|
Characteristics |
Greater genomic stability High fidelity DNA polymerases Larger genomes Greater genomic packaging capacity May or may not replicate in presence of nAbs Longer replication duration Nuclear Replication Mechanisms to block DNA virus sensing adaptors |
Genomic instability Low-fidelity RNA polymerase Smaller genomes Limited genomic packaging capacity Shorter replication duration Cytoplasmic replication Rapid evolution Mechanisms to block RNA virus sensing adaptors |
Family |
Adenoviridae / Adenovirus Herpesviridae / Herpes Simplex 1 virus Parvoviridae / Parvovirus H1 virus Poxviridae / Myxoma Vaccinia virus |
Orthomyxoviridae / Influenza A Echovirus, Coronaviridae Paramyxoviridae / Measles virus Paramyxoviridae / Mumps virus Paramyxoviridae / NDV Reoviridae / Reovirus Picornaviridae / Poliovirus Picornaviridae / Seneca Valley Virus Rhabdoviridae / Vesicular Stomatitis virus Togavirdae / Semliki forest Sindbis virus Retroviridae / Moloney Murine Leukemia |
Cancer Targets |
Head and Neck, Breast, Prostate, Ovarian, Osteosarcoma, Colon, etc. |
Melanoma, Multiple Myeloma, Glioma, Lymphoma, Pancreatic adenocarcinoma, mesothelioma, etc. |
Today, OVs have been used in clinical trials for treatment and there is an OV product, T-VEC, that has been approved by the FDA. Henceforth, oncolytic virotherapy (featuring: specificity for target cancer; high-efficiency to kill infected cells; and safety to avoid adverse reactions) has been regarded as a promising and effective approach for cancer therapeutics that is easier to destroy tumor cells compared with radiation therapy and chemotherapy. Approved and novel OV products are discussed next.
4 INNOVATIVE ONCOLYTIC VIROTHERAPY AND APPROVED OVS
The field of oncolytic virotherapy is continually gaining prominence as a viable option for novel cancer immunotherapy, and robust developmental pipelines have resulted in the approval of four OVs worldwide. The initial registered OV, ECHO-7 (commercially known as Rigvir-trade name), received approval in Latvia in 200[75]. ECHO-7, a type 7, group IV, enteric cytopathogenic human orphan (ECHO) virus, underwent multiple passages in human tumor tissue cultures and was selected for enhanced selective replication within tumor cells[75,76]. Initially approved for the local treatment of skin and subcutaneous melanoma metastases via intramuscular injections, ECHO-7 has demonstrated effectiveness across various cancer types, including colorectal, gastric, and small cell lung cancers[77,78]. Pumpure and colleagues reported in 2020 the treatment of a female patient diagnosed with stage IV primary malignant melanoma of the cervix, noting no side effects or adverse reactions. At the time of publication, the patient had achieved a survival of 67 months and a progression-free survival (PFS) of 57 months[79]. However, the State Agency of Medicines of Latvia suspended the marketing authorization of Rigvir in 2019 due to issues related to poor quality control[80]. In 2005, the Chinese State Food and Drug Administration granted approval for H101 (marketed with the trade name Oncorine) to treat head and neck cancer[81]. H101 is a type 5 recombinant human adenovirus featuring deletions in the gene encoding the 55kDa E1B protein and the E3 region gene segment. The removal of the E1B gene facilitates proper p53 tetramer formation and regulation of the cell cycle checkpoint, as E1B typically binds and inactivates[82]. The E3 region, with its seven expressed open reading frames, plays a role in inhibiting host immunity to enhance viral dissemination[83]. Extensive testing has been conducted on H101 for various solid tumors, including gastric carcinoma, HCC, and lung cancer[84-86]. In 2021, a study by Zhang et al.[86] investigated H101 treatment, with or without chemotherapy, in 95 patients diagnosed with advanced gastric cancer. The findings indicated that the combination therapy with H101 was more effective, resulting in a median overall survival (OS) of 29 months and a median PFS of 14.8 months, compared to single-agent H101 or chemotherapy alone[86]. In 2015, talimogene laherparepvec (trade name T-VEC) marked a significant milestone as the first oncolytic virus approved by the FDA for the local treatment of unresectable cutaneous, subcutaneous, and nodal lesions associated with advanced melanoma or postoperative recurrent melanoma. T-VEC, a genetically modified herpes simplex 1 virus (HSV-1), underwent deletion of both copies of the gene encoding infected cell protein 34.5 (ICP34.5), a peptide enhancing the virus’ neurovirulence[87]. Subsequently, these deletions were replaced with a gene encoding GM-CSF. This genetic alteration induces the secretion of GM-CSF, promoting the recruitment of antigen-presenting cells (APCs) to the TME and enhancing cytotoxic T lymphocyte (CD8+ T-cell) responses to tumor-associated antigens (TAAs). This modification is believed to enhance viral replication in tumor cells with defective IFN pathways[88-91]. Primarily implemented in melanoma treatment, T-VEC has also undergone clinical trials focused on lymphomas[92,93]. In a 2021 study by Ramelyte et al.[88], intralesional T-VEC treatment of 13 patients with primary cutaneous B cell lymphomas (pCBCL) demonstrated mild side effects such as flu-like symptoms. No patients developed suspected HSV-associated systemic infection. T-VEC treatment showcased enhanced recruitment of an early innate immune response, including activated NK cells and monocytes, followed by increased CD8+ T-cell populations and reduced T regulatory cell (Treg) populations.
Overall, T-VEC treatment proved effective in treating pCBCL, with a complete response (CR) of 46.2%, partial response (PR) of 38.4%, and progressive disease of 15.4%[88]. In a 2016 phase IB trial investigating T-VEC in combination with Ipilimumab, a CTLA-4 inhibitor, in 19 patients with stage IIIB-IVM1c melanoma deemed unsuitable for surgical resection, Puzanov et al.[94] reported the combination treatment as safe. Some patients experienced grade 3/4 adverse events, but these did not result in discontinuation of T-VEC or ipilimumab. The treatment exhibited promising results, with a CR of 22%, PR of 28%, and stable disease of 22%. The probability of survival at 12 months and 18 months was 72% and 67%, respectively[94]. Harrington et al.[95] conducted a phase III OPTiM trial in 2016 comparing the response rate of intra-tumoral injection of T-VEC to subcutaneous injection of GM-CSF in 249 patients with stage IIIB/C or IVM1a melanoma. The oncolytic virus treatment (Durable Response Rate (DRR)=25.2%, Overall Response Rate (ORR)=40.5%) was found to be more beneficial than GM-CSF treatment (DRR=25.2%, ORR=2.3%). The median OS of T-VEC versus GM-CSF treatment was 41.1 and 21.5 months, respectively. Both therapeutic arms were well-tolerated, with patients reporting mild adverse events such as chills, fatigue, and influenza-like illness[95]. Consequently, the data presents encouraging results, warranting further in-depth research to confirm these findings. In 2021, Teserpaturev (or Teserpaturev/G47Δ with the trade name DELTACT) received conditional approval for the treatment of malignant glioma in Japan. Teserpaturev, an HSV-1 variant, features the deletion of both copies of the γ34.5 gene and the α47 gene, with the US11 promoter. Additionally, the lacZ gene was inserted to inactivate the ICP6 gene[96]. The γ34.5 gene, responsible for inhibiting the host cell-induced shutdown of protein synthesis in response to viral infection, was deleted to enable viral replication in cancer cells, given that malignant cells often lack the ability to halt protein synthesis[97]. Removal of the α47 gene eliminates viral inhibition of host cell transporters associated with antigen presentation, enhancing anti-tumor immune activation[98]. Furthermore, inactivation of the ICP6 gene induces selective viral replication in actively dividing cells, as ICP6 encodes the large subunit of ribonucleotide reductase necessary for viral DNA replication[99]. In a 2021 study conducted by Uchihashi et al.[100], Teserpaturev/G47Δ’s efficacy in treating oral squamous cell carcinoma was investigated using a murine model. Teserpaturev / G47Δ was observed to inhibit the growth of primary lesions and prolong the survival of athymic nude and immunocompetent mice injected with tongue cancer cells. Injected Teserpaturev/G47Δ demonstrated immediate dissemination into cervical lymph nodes, effectively suppressing lymph node metastases[100].
5 LIMITATIONS AND CHALLENGES OF CONTEMPORARY OVS THERAPY
Introducing and / or implementing OVs into our clinical practice demands meticulous consideration, given the array of factors influencing their effectiveness. The choice of inoculation methods entails weighing the advantages and disadvantages associated with viral therapy. Additionally, careful attention must be directed towards the tumor extracellular matrix (ECM) and the intricate tumor stroma, recognizing their significance in oncolytic virotherapy. Notably, cell populations such as cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs) can significantly impede the efficacy of oncolytic virotherapy.
5.1 Administration-related Challenges for OVs
OVs can be introduced through either direct inoculation into the tumor bulk or systemic injection, encompassing intravenous or intra-arterial administrations[101]. Each administration method carries distinct advantages and challenges (Table 2). Direct intra-tumoral inoculation, exemplified by the FDA fast-tracking of T-VEC, has demonstrated notable success by concentrating the virus at the lesion site, eliciting a robust immunological response. However, this approach is constrained by limited tumor accessibility, particularly for deep-seated or sensitive location tumors, where invasive procedures pose risks of injury and complications. Challenges also include poor intra-tumoral retention due to viral dissemination into the bloodstream, limited dispersion within tumor tissues, and potential inflammatory responses[102]. Conversely, systemic therapy utilizes the vascular system of the body to disseminate OVs throughout, akin to the delivery of chemotherapy or other anti-cancer agents. Despite its advantages, indirect inoculation presents potential drawbacks. Concerns center around systemic toxicity, questioning whether the dosage of OVs might inadvertently cause off-tumor tissue or organ damage. Another critical consideration is immune clearance, involving neutralization of OVs by B cell-generated antibodies, impeding virus internalization and substantially reducing the viral titer reaching the tumor site[101,103]. Herein, this raises concerns regarding the sero-positivity (i.e., having a +Ve serum reaction in an antibody test) to the backbone virus, particularly crucial for viruses highly prevalent in the community. Human adenovirus (hAdV) infections exhibit high seropositivity rates worldwide, including in the, United States, Australia, Japan, and the Philippines. For instance, a 2018 study by Ye et al.[104] assessed nAbs prevalence against HAdV types 4 and 7 in volunteers from Hunan Province in China, revealing seropositivity rates of 58.4% for HAdV4 and 63.8% for HAdV. Consequently, it can be anticipated that a substantial portion of global populations in regions with a history of HAdV infection maintains high seropositivity for HAdV nAbs. The challenge persists in that seropositivity constrains viral replication[105]. nAbs bind to OVs, impeding cellular receptor binding[103,106]. Hence, wary decisions regarding the viral strain and administration method should be meticulously considered in light of pre-existing immune responses.
To the best of knowledge, the consensus in the literature emphasizes the necessity of suppressing humoral immunity for systemically administered oncolytic virotherapy[107]. The IFN pathway, particularly IFN-α, acts as an adversary to OVs by diminishing viral replication and impeding virus-mediated apoptosis[108]. Given that cancer cells often lack a type 1 IFN response, they exhibit greater permissiveness to OV infection and replication[59]. Efforts have been undertaken to shield OVs from the innate and adaptive immune system, specifically the humoral response, using IFN response inhibitors to augment viral replication and enhance oncolysis efficacy. Nevertheless, concerns about the safety of IFN antagonists have been raised. Saren et al.[109] observed in 2017 that treating glioblastoma-bearing mice with Semliki Forest virus equipped with vaccinia virus-encoded type 1 IFN decoy receptor B18R effectively controlled tumor growth but also triggered severe neurotoxicity as the virus disseminated and replicated into/in the healthy brain tissue.
An alternative approach to protect OVs involves employing genetically engineered protective coatings, including chemical polymers, cell-derived nanovesicles, and liposomes. These coatings offer a direct means of overcoming the humoral immune response[110-112], diminishing immune recognition of the virus and thereby limiting the production of nAbs against the OVs. Introducing tumor-targeting ligands can enhance the homing capabilities of OVs toward the tumor. However, a major concern with protective coatings lies in their practicality. While they increase the viral titer reaching the tumor, these coatings may compromise the ligand-receptor interactions between OVs and tumor cell receptors, resulting in reduced OV internalization. Additionally, challenges include high production costs and limitations in large-scale OV transport[107]. Another potentially viable protective method involves using carriers, such as patient-derived cellular carriers (e.g., OV-infected cells injected back into the patient) or engineered carriers (e.g., nanoparticles). Various cell types, including endothelial cells, mesenchymal stromal cells (MSCs), T-cells, and even tumor cells, can serve as cellular carriers. However, safety concerns arise with certain cell types, necessitating thorough safety assessments, even when using the patient’s own tumor cells. MSCs or neuronal stem cells, known for tumor tropism, allow OV delivery throughout the body but also pose challenges, given their ability to evade the immune system and facilitate immune escape of tumor cells. The utilization of biodegradable nanoparticles is gaining popularity for efficiently delivering viral antigens. The diverse selection of nonmetal and metal-based compositions enhances the delivery of OVs[107], with liposomal nanoparticles emerging as a promising carrier due to their high biocompatibility and rapid degradation by macrophages, making them favorable for OV transport[113]. Lipid-based core-shell nanocapsules (comrprising layer-by-layer self-assembled natural biopolymers) are being formulated in our BioMAT’X R&D&I (HAiDAR I+D+i) laboratory, at CiiB- UAndes in Chile, for the controlled delivery of OVs.
Table 2. Anti-cancer Properties and Challenges in Delivery of OVs
OVs as an Anti-cancer Drug Class |
Challenges in Delivery of OVs |
Viruses naturally favor cancer cell infection. Natural affinity for tumor tissues. |
OVs deviate from conventional pharmacological principles (biological amplification). |
Directed elimination of cancer cells. Genetic engineering enhances properties. |
Intravenous delivery allows virus circulation to distant metastatic sites. |
OVs are emerging as a new anticancer drug class. |
Extravasation into the tumor parenchyma is inefficient. |
OVs target tumor tissues, kill tumor cells, and amplify anti-tumor immunity. |
Intra-tumoral injection concentrates the virus at the tumor site. |
Safety for patients and involved healthcare staff is crucial. |
Regression of distant tumors requires systemic spread or immune response induction. |
Diversity in virus families and engineering for customized and personalized OVs. |
Challenges include nAbs, macrophage sequestration, and virus dilution. |
5.2 Challenges Arising from Tumor Structure for OVs
Beyond the cellular level, physical impediments such as tumor stroma pose challenges for chemotherapy, tumor-infiltrating effector cells, and OVs in reaching tumor cells effectively[114,115]. Comprising non-tumor cells and structural components, the tumor stroma acts as a barrier to immune infiltration. Tumor cells release cytokines to suppress anti-tumor immune functions, while stromal cells construct a desmoplastic stroma barrier, further hindering immune penetration[116]. Encompassing the dense ECM, CAFs, TAMs, and tumor vasculature, the stroma collectively reinforces the tumor’s resistance to the host’s immune system[117-119]. The ECM, primarily generated by CAFs, presents a significant barrier due to its substantial mass, forming an impenetrable shield around the tumor that compromises immune invasion and the efficacy of anti-tumor drugs[119]. The ECM’s density also contributes to a scarcity of oxygen and nutrients, exploited by tumor cells to activate metabolic stress-related signaling pathways, allowing them to shape the TME to their advantage. For instance, vascular endothelial cells (VECs) can transform into tumor endothelial cells (TECs), exhibiting enhanced proliferation, increased migration, and facilitation of angiogenesis[120,121]. Additionally, the ECM’s dense structure activates drug efflux pumps and induces senescence, enhancing tumor resistance against anti-cancer agents like chemotherapy[119]. CAFs recruit myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), fostering an immunosuppressive environment[122]. M2-type macrophages, with an anti-inflammatory profile, secrete TGF-β, promoting collagen secretion and cross-linking, reinforcing the ECM[114,123]. Strategies to target tumor stroma include OVs expressing proteases like matrix metalloproteinase (MMP)-9 to degrade ECM components. For instance, Sette et al.[124] (2019) demonstrated that treating glioblastoma multiforme with MMP-9-armed HSV-derived OVs increased viral invasion and improved survival in tumor-bearing nude mice. OVs can also be equipped with tissue inhibitor metalloproteinases 1-4 (TIMPs 1-4) to regulate MMPs’ proteolytic activity and prevent ECM rearrangement[125]. Another approach involves repolarizing M2-type macrophages into the pro-inflammatory M1-type phenotype[126,127], which secretes cytokines and reactive oxygen species to enhance immune recruitment and function against malignant cells[128]. In 2020, Rao et al.[129] showcased the use of genetically engineered cell membrane-coated magnetic nanoparticles triggering M2 to M1 repolarization, inhibiting tumor proliferation, reducing metastasis, and improving survival in mice with triple-negative breast cancer. While OVs can influence the TME to shift it from a pro-tumor to an anti-tumor environment, there remains room for improvement in the strategies described above. Figure 2 provides an overview on how OVs work to overcome resistance to immuno-therapy as well as underscores the urging need for innovative approaches in OVs development, emphasizing further research in targeting both the tumor and its surrounding stroma.
|
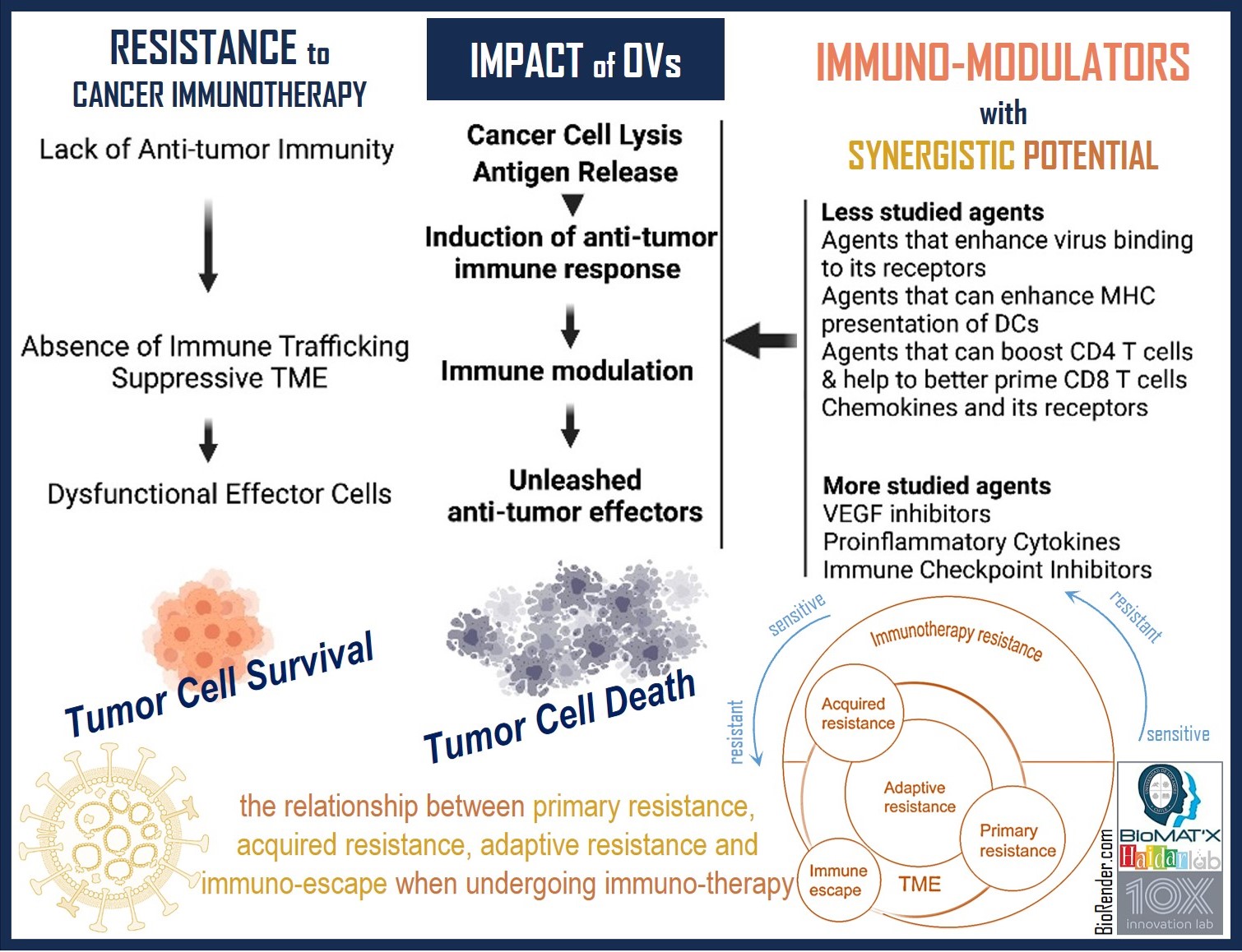
Figure 2. Resistance to Immunotherapy through Emerging OVs. The resistance mechanisms to existing immune checkpoint inhibitors and other immuno-therapies are diverse and multi-factorial. OVs offer a promising avenue to overcome primary or secondary resistance to immuno-therapy, either as standalone agents or in conjunction with other immuno-modulating compounds. OVs as a promising strategy to overcome resistance to immuno-therapy, works, as follows: (1) Direct Tumor Cell Lysis: OVs are designed to selectively infect and kill cancer cells. By directly targeting tumor cells, OVs can induce tumor cell death, releasing tumor-specific antigens into the TME. (2) Immunogenic Cell Death (ICD): Some OVs induce ICD, a form of cell death that stimulates the immune system. This process can lead to the release of DAMPs and TAAs, which activate dendritic cells and prime T cells for an anti-tumor response. (3) Innate Immune Stimulation: OVs can also activate innate immune responses by inducing the production of pro-inflammatory cytokines and chemokines, recruiting immune cells such as macrophages, natural killer (NK) cells, and dendritic cells to the tumor site. (4) Breaking Immune Suppression: OVs can counteract immunosuppressive mechanisms within the TME. For example, they can target and kill regulatory T cells (Tregs) or MDSCs, which suppress anti-tumor immune responses. (5) Combination Therapy: OVs can be used in combination with other immuno-modulating compounds, such as immune checkpoint inhibitors (ICIs), to enhance their efficacy. By combining OVs with ICIs, researchers aim to overcome resistance mechanisms that may limit the effectiveness of either treatment alone. (6) TME Modification: OVs have the potential to modify the TME, making it more conducive to anti-tumor immune responses. For example, OVs can induce vascular normalization, reduce tumor hypoxia, and remodel the ECM, which can enhance the infiltration and function of immune cells within the tumor. By leveraging these mechanisms, OVs offer a promising approach to overcome primary or secondary resistance to immuno-therapy and enhance the effectiveness of cancer treatment.
6 OVS IN CLINICAL TRIALS
Considering the collective advancements in oncolytic virotherapy, numerous active clinical trials are currently exploring the efficacy of OVs across a spectrum of cancers, including breast, gastrointestinal, skin, and pancreatic cancers. Prominent OV candidates in these trials encompass VV, HSV, and adenovirus. While some clinical trials exclusively assess patient responses to single-agent OV therapy, the majority adopt a combination approach, often integrating OV treatment with chemotherapy, monoclonal antibodies, or radiotherapy (Table 3, presents some of the ensuing data retrieved from clinicaltrials.gov in 05/2022). However, there exists a crucial necessity for studies identifying accurate biomarkers to customize and optimize the therapeutic strategy(ies) that blend various treatments for the specific patient, recognizing the potential variations in disease characteristics among the individuals.
Table 3. Selected Clinical Trials Involving OVs and other Anti-cancer Therapies in a Variety of Routes and Applications
Clinical Trial ID |
Tumor |
Phase |
OVs |
Administration Route |
Co-Treatment/Therapy |
NCT02705196 |
Pancreatic cancer |
I / II |
ADV |
Intra-tumoral |
Nucleoside, anti-PD1 Ab, & Anti-microtubule agent |
NCT03004183 |
NSCLC & breast cancer |
II |
ADV / HSV |
Intra-tumoral |
nucleoside, radiation, & anti-PD1 Ab |
NCT03916510 |
Rectal cancer |
I |
ADV |
Intravenous |
Radiotherapy & antimetabolite |
NCT05051696 |
FG neoplasms |
NA |
ADV |
Intra-tumoral |
Radiotherapy |
NCT05234905 |
FG neoplasms |
II |
ADV |
Intra-tumoral |
anti-PD1 Ab |
NCT03252808 |
Pancreatic cancer |
I |
HSV |
Intra-tumoral |
Nucleoside & Anti-microtubule agent |
NCT03663712 |
Ovarian cancer |
I |
HSV |
Intraperitoneal |
NA |
NCT03866525 |
GIC |
I / II |
HSV |
Intra-tumoral |
TOP1 inhibitor & anti-PD1 Ab |
NCT04050436 |
SCSC |
II |
HSV |
Intra-tumoral |
anti-PD1 Ab |
NCT04185311 |
Breast cancer |
I |
HSV |
Intra-tumoral |
anti-PD1 Ab, anti-CTLA4 Ab |
NCT04349436 |
Carcinoma |
I / II |
HSV |
Intra-tumoral |
NA |
NCT04755543 |
GIC |
I |
HSV |
Intravenous |
anti-PD1 Ab, alkylating agent, anti-metabolites |
NCT05232136 |
Bladder cancer |
I / II |
HSV |
Intravesical |
NA |
NCT05235074 |
CNS tumors |
I / II |
HSV |
Intra-tumoral |
NA |
NCT03043391 |
Glioma |
I |
Poliovirus |
Intra-tumoral |
NA |
NCT03564782 |
Breast cancer |
I |
Poliovirus |
Intra-tumoral |
NA |
NCT04445844 |
Breast cancer |
II |
Reovirus |
Intravenous |
anti-PD-L1 Ab |
NCT02977156 |
Advanced cancer |
I |
VV |
Intra-tumoral |
anti-CTLA4 Ab |
NCT03206073 |
CRC |
I / II |
VV |
Intravenous |
anti-CTLA4 Ab, anti-PD-L1 Ab |
NCT03954067 |
Advanced cancer |
I / II |
VV |
Intra-tumoral |
w / wo anti-PD1 Ab |
NCT04787003 |
Advanced cancer |
I |
VV |
Intra-tumoral |
w / wo anti-PD1 Ab, anti-PD-L1 Ab |
Notes: Abbreviations: adenovirus (ADV), non-small cell lung cancer (NSCLC), herpes simplex virus (HSV), female genital (FG), gastrointestinal cancer (GIC), Topoisomerase 1 (TOP1), squamous cell skin cancer (SCSC), central nervous system (CNS), colorectal cancer (CRC), and Vaccinia virus (VV).
Table 3 above provides an overview of the expanding research on utilizing immune checkpoint inhibition (ICI) in conjunction with OVs to eliminate tumor cells[11,48]. Numerous monoclonal antibodies have been designed to target key immune checkpoints, including cytotoxic T-lymphocyte antigen 4 (CTLA-4), PD-1, and programmed death protein ligand 1 (PD-L1). PD-1 plays a crucial role in sustaining exhausted T cells, and inhibiting PD-1 post-exhaustion can enhance T cell immune effector functions, disrupting the immune evasion employed by tumor cells[130]. The advent of ICI, marked by the FDA approval of the anti-CTLA antibody ipilimumab in 2011, has transformed the landscape of cancer care. However, the ORR remains limited, typically ranging from 20-40% in most studied regimens. Consequently, addressing primary resistance to ICI by fostering the generation of novel tumor antigen-specific immune responses and innate immune responses, while simultaneously shifting the TME towards a pro-inflammatory state, holds significant promise. In 2017, Ribas et al.[22] examined the impact of combining oncolytic virotherapy (T-VEC) with pembrolizumab, an anti-PD-1 antibody, in patients with advanced melanoma. Earlier research had indicated that some patients exhibit resistance to PD-1 blockade due to the scarcity of CD8+ T cells within the tumor lesion[131,132]. The co-administration of T-VEC and anti-PD1 blockade induced a robust immune response, augmenting the systemic circulation of CD4+ and CD8+ T cells, elevating T cell infiltration into tumors, and mitigating T cell exhaustion. Common T cell inhibitory markers, including CTLA4, PD-1, TIGIT, TIM3, and LAG3, exhibited reduced expression[133]. In a phase 1b study (n=21), this combination treatment demonstrated a tumor size reduction, achieving an ORR of 62% and a CR rate of 33%, with low toxicity[22]. However, the subsequent phase 2 study (n=692) conducted in a similar setting showed an acceptable safety profile but did not meet the primary endpoint for PFS at 14.3 months (median; range=10.3-22.1), while the placebo and pembrolizumab arm demonstrated a PFS of 8.5 months (median; range=5.7-13.5, hazard ratio=0.86; CI=0.71-1.04, p=0.13). The OS, as part of the dual primary endpoint strategy, is yet to be reported[134]. While this approach demonstrated feasibility, further investigation is necessary to identify the most effective and synergistic combination regimen, along with predictive biomarkers, to enhance the anti-tumor activity and minimize unnecessary adverse events for select patients who would benefit most from the treatment.
OVs in clinical trials show distinct advantages compared to traditional therapies such as chemotherapy and radiation therapy, particularly in terms of OS, PFS, and disease-free survival (DFS). OVs selectively infect and kill cancer cells while sparing normal tissue, offering a targeted approach that minimizes systemic toxicity. In clinical trials, OVs have demonstrated the potential to improve OS and PFS by not only directly lysing tumor cells but also by triggering a robust immune response that can target both primary and metastatic sites. This dual mechanism could also enhance DFS by reducing the likelihood of disease recurrence. Unlike chemotherapy, which can lead to widespread side effects and resistance, OVs provide a more targeted therapy with a favorable safety profile. Compared to radiation therapy, OVs offer systemic efficacy, which is particularly beneficial in treating metastatic or recurrent disease, where localized treatments may fall short. To recap, OVs in clinical trials, to date, and to the best of knowledge, are showing promise in improving survival outcomes, particularly in cancers where traditional therapies have limited effectiveness.
7 OVS DELIVERY AND PHARMACOO DYNAMICS
As noted earlier in this review, the safe and effective delivery of OVs poses a significant challenge in the field of oncology, limiting their therapeutic impact and potential. Also among the over 3,000 virus species, not all are suitable for use as oncolytic agents. This may contribute to the observed differences between the impressive in vitro and in vivo pre-clinical studies[135,136] and the relatively modest anti-tumor effects observed in clinical trials thus far[137], emphasizing further the need for more potent yet safe treatment strategies (for long-term tumor control, in human patients). Generally, OVs must be non-pathogenic and possess inherent cancer-selective killing activity or be engineered to express attenuating genes or arming genes. Through genetic engineering, it is possible today to design live replicating viruses that exhibit high tumor selectivity by targeting cell entry and transcription. Additionally, these engineered viruses can be armed with reporter genes for non-invasive monitoring of virotherapy pharmaco-kinetics and -dynamics, as well as for enhancing cytotoxic activity, promoting ICD, or modulating the immune response. Commercially-available OVs for cancer treatment such as Rigvir, Oncorine H101 (in combination with chemotherapy), and talimogene laherparepvec or T-VEC were discussed earlier, and continue to suffer limitations and challenges. Monotherapies capable of significantly impacting the OS of cancer patients are desirable. Furthermore, it is worth remembering that a key differentiator between OVs and conventional drugs is their ability to self-amplify and disseminate post-delivery, potentially delaying the attainment of peak concentration until sometime after treatment administration. Hence, to enhance delivery, and attain a crucial viral concentration within the tumor (intra-lesional / -tumoral approach is favored, when compared to systemic routes, mainly owing to reduced viral inactivation by the innate immune system, a lower risk of systemic toxicity, and the efficient delivery of a substantial viral load in a single dose; and deemed essential to enable substantial oncolysis and the safe, reproducible systemic spread of the virus to all disease sites), it is believed and proposed that 3 key limitations must be addressed: (1) the bioavailability of the virus and achieving therapeutic levels, influenced by host vascular dynamics, perfusion parameters, and innate immune responses[138-140]; (2) bio-distribution and propagation of OVs[141-143], often hindered by the heterogeneity of the intra-tumoral microenvironment and heterogeneous ECM; and (3) the amplification of the by-stander killing effect of the virus through cell-to-cell contact or intrinsic vector enhancement[139]. Herein, the combination of OVs with MSCs presents a promising and targeted approach to cancer treatment. Leveraging the migratory and tumor-targeting properties of MSCs, along with the tumor cell-destroying abilities of OVs, makes this strategy suitable for both localized and metastatic malignancies. Moreover, the capacity of OVs to induce ICD and trigger anti-tumor immune responses enhances the therapeutic potential. Importantly, the combination of MSCs and OVs also provides an opportunity to modulate the TME, thereby augmenting the effectiveness of immuno-therapies[143-145]. However, whether shielding is done by cell- or nanoparticle-based delivery approaches (passive versus active delivery), as afore-mentioned, several challenges continue to need addressing, including the optimization of delivery procedures, using biodegradable biomaterials and encapsulation coatings with superior physico-chemico-mechanical properties, improvement of safety profiles, and enhancement of OV efficiency[143-145]. Further studies are necessary to determine the optimal timing, dosage, and frequency of administering carrier-releasing OVs in different cancer types and stages. Given the ongoing progress in oncolytic virotherapy R&D&I, there is irrefutable evidence that OVs are poised as fundamental components of future cancer treatment protocols, with the promising potential to transform the paradigm of cancer care and patient quality of life.
OVs offer a promising approach to treating metastatic cancer by targeting cancer cells through mechanisms independent of specific tumor markers, which often change during metastasis. They can overcome resistance to conventional therapies by inducing direct oncolysis and stimulating a systemic anti-tumor immune response, effectively targeting both active and dormant cancer cells. Additionally, OVs can enhance the efficacy of combination therapies, rendering them a versatile and powerful tool against the challenges of metastatic disease, such as tumor heterogeneity and therapy resistance, and beyond.
8 CONCLUSIONS AND FUTURE DIRECTIONS
Cancer remains a major health problem and a leading cause of death World-wide. Indeed, it is becoming increasingly harder to treat, with more patients now resistant to chemotherapies. So, with around 10 million cancer patients suffering and dying each year, OVs have emerged as a new class of cancer therapeutics and a prominent player in immuno-therapy, providing a diverse array of viruses that can be genetically modified to specifically target and replicate within tumor cells, sparing normal cells from harm. The process of cell lysis not only releases various factors that attract immune cells to the tumor but also allows viral progeny to infect neighboring tumor cells, perpetuating the oncolytic cycle. The conditional and selective replication of OVs alongside their broad immune-stimulatory effect (ability to stimulate the host adaptive immune response) makes them an attractive therapeutic option. However, the successful (safe and efficacious) administration of OVs faces challenges, including viral neutralization by the humoral immune response and the hostile TME, necessitating further investigation. Some studies have explored the use of protective coatings or cellular carriers to overcome viral neutralization and enhance the delivery of OVs to the tumor site, yet with ongoing limitations and challenges as well. Indeed, while the prospects of MSC-releasing OVs appear promising, their efficacy and safety must be validated through additional research and clinical trials. Such a comprehensive evaluation of genetically-engineered OVs, virotherapy, delivery systems, including MSCs and nanoparticles, amongst others, underscores the potential benefits of OVs delivery strategies and pharmacodynamics while emphasizing the importance of ongoing investigation and innovation in this rapidly evolving field. Finally, a review of the ongoing clinical trials in oncolytic virotherapy, as documented in the available and accessed clinical trial registries, underscores the profound interest within the biomedical community, particularly in combination approaches with conventional cancer treatments such as surgery, chemotherapy, and radiation, as well as novel immune modulators. Future studies must confirm the long-term safety and efficacy of incorporating OVs therapy. Additionally, research efforts should focus on developing strategies that target cancer heterogeneity while ensuring proper receptor binding for viral entry in the face of rapidly evolving cancer cells. Precision medicine may play a crucial role in offering a personalized approach for patients. In summary, oncolytic virotherapy has solidified its position as the fourth pillar of cancer treatment, supporting cancer immuno-therapy, and ongoing R&D&I will continue to explore the diverse application(s) of OVs in multi-modal and -factorial clinical approaches.
Acknowledgments
This work was supported by operating grants provided to the HAiDAR I+D+i/R&D&I LAB/BioMAT’X (Laboratory of Biomaterials, Pharmaceuticals and Cranio-Maxillofacial Tissue Bioengineering), member of CiiB (Biomedical Research and Innovation Center), Faculties of Medicine and Dentistry, Universidad de los Andes, Santiago de Chile, through the ANID-NAM (National Research and Development Agency, Chile and National Academy of Medicine, NIH, USA) Grant Code # NAM21I0022 (2020-2022), CORFO Creaty Valida I+D+i Grant Code # 21CVC2-183649 (2021-2023), CORFO Creates and Validates-Collaborative R&D&I Project-Reactivate” Grant Code # 22CVC2-218196 (2022-2024), and FONDEF Concurso IDEA de I+D, ANID, Grant Code # ID22I10215 (2022-2024). The author wishes to acknowledge Dr. Karina Pino, Immunology Group at CiiB-UAndes for her invaluable insights in improving this review article.
Conflicts of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data Availability
No additional data are available.
Copyright Permissions
Copyright © 2024 The Author(s). Published by Innovation Forever Publishing Group Limited. This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.
Author Contribution
The author solely contributed to draft the manuscript and approved the final version.
Abbreviation List
CAFs, Cancer-associated fibroblasts
CR, Complete response
DAMPs, Damage-associated molecular patterns
DFS, Disease-free survival
DRR, Durable Response Rate
DsDNA, Double-stranded DNA
ECHO, Enteric cytopathogenic human orphan
ECM, Extracellular matrix
FDA, The US Food and Drug Administration
GM-CSF, Granulocyte-macrophage colony-stimulating factor
HAdV, Human adenovirus
HCC, Hepatocellular carcinoma
HSV, Herpes simplex virus
ICD, Immunogenic cell death
ICI, Immune checkpoint inhibition
IFN, Interferon
MDSCs, Myeloid-derived suppressor cells
MSCs, Mesenchymal stromal cells
NAbs, Neutralizing antibodies
NDV, Newcastle disease virus
ORR, Overall Response Rate
OS, Overall survival
OVs, Oncolytic viruses
PAMPs, Pathogen-associated molecular patterns
Pcbcl, Primary cutaneous B cell lymphomas
PD-1, programmed death protein 1
PD-L, programmed death protein igand 1
PFS, Progression-free survival
PR, Partial response
Rb, Retinoblastoma
RD, Replication-defective
TA, Tumor antigens
TAAs, Tumor-associated antigens
TAMs, Tumor-associated macrophages
TECs, Tumor endothelial cells
Treg, T regulatory cell
TK, Thymidine kinase
TME, Tumor microenvironment
TNF, Tumor necrotic factor
T-VEC, Talimogene laherparepvec
VECs, Vascular endothelial cells
VV, Vaccinia virus
References
[1] Sung H, Ferlay J, Siegel RL et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca-Cancer J Clin, 2021; 71: 209-249.[DOI]
[2] Pulendran B, Davis MM. The science and medicine of human immunology. Science, 2020; 369: eaay4014.[DOI]
[3] Monath TP. Yellow fever vaccine. Expert Rev Vaccines, 2005; 4: 553-574.[DOI]
[4] Vetter V, Denizer G, Friedland LR et al. Understanding modern-day vaccines: What you need to know. Ann Med, 2018; 50: 110-120.[DOI]
[5] Raman SS, Hecht JR, Chan E. Talimogene laherparepvec: review of its mechanism of action and clinical efficacy and safety. Immunotherapy, 2019; 11: 705-723.[DOI]
[6] Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol, 2020; 13: 84.[DOI]
[7] Goswami R, Subramanian G, Silayeva L et al. Gene Therapy Leaves a Vicious Cycle. Front Oncol, 2019; 9: 297.[DOI]
[8] Duerner LJ, Schwantes A, Schneider IC et al. Cell entry targeting restricts biodistribution of replication-competent retroviruses to tumour tissue. Gene Ther, 2008; 15: 1500-1510.[DOI]
[9] Anderson BD, Nakamura T, Russell SJ et al. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res, 2004; 64: 4919-4926.[DOI]
[10] He Y, Mueller S, Chipman PR et al. Complexes of poliovirus serotypes with their common cellular receptor, CD155. J Virol, 2003; 77: 4827-4835.[DOI]
[11] Rahman MM, McFadden G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers, 2021; 13: 5452.[DOI]
[12] Borrego-Diaz E, Mathew R, Hawkinson D et al. Pro-oncogenic cell signaling machinery as a target for oncolytic viruses. Curr Pharm Biotechnol, 2012; 13: 1742-1749.[DOI]
[13] Matveeva OV, Chumakov PM. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev Med Virol, 2018; 28: e2008.[DOI]
[14] De Munck J, Binks A, McNeish IA et al. Oncolytic virus-induced cell death and immunity: a match made in heaven? J Leukoc Biol, 2017; 102: 631-643.[DOI]
[15] Kroemer G, Galluzzi L, Kepp O et al. Immunogenic cell death in cancer therapy. Annu Rev Immunol, 2013; 31: 51-72.[DOI]
[16] Tang D, Kang R, Coyne CB et al. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol Rev, 2012; 249: 158-175.[DOI]
[17] Krysko DV, Garg AD, Kaczmarek A et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer, 2012; 12: 860-875.[DOI]
[18] Hua L, Wakimoto H. Oncolytic herpes simplex virus therapy for malignant glioma: Current approaches to successful clinical application. Expert Opin Biol Ther, 2019; 19: 845-854.[DOI]
[19] Davola ME, Mossman KL. Oncolytic viruses: how “lytic” must they be for therapeutic efficacy? Oncoimmunology, 2019; 8: e1581528.[DOI]
[20] Li Z, Jiang Z, Zhang Y et al. Efficacy and Safety of Oncolytic Viruses in Randomized Controlled Trials: A Systematic Review and Meta-Analysis. Cancers, 2020; 12: 1416.[DOI]
[21] Bonaventura P, Shekarian T, Alcazer V et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol, 2019; 10: 168.[DOI]
[22] Ribas A, Dummer R, Puzanov I et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell, 2017; 170: 1109-1119.e10.[DOI]
[23] Barry M. Single-cycle adenovirus vectors in the current vaccine landscape. Expert Rev Vaccines, 2018; 17: 163-173.[DOI]
[24] Ledgerwood JE, Sullivan NJ, Graham BS. Chimpanzee Adenovirus Vector Ebola Vaccine--Preliminary Report. N Engl J Med, 2015; 373: 776.[DOI]
[25] Harrington K, Freeman DJ, Kelly B et al. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov, 2019; 18: 689-706.[DOI]
[26] Lawler SE, Speranza MC, Cho CF et al. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol, 2017; 3: 841-849.[DOI]
[27] Beachboard DC, Horner SM. Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol, 2016; 32: 113-119.[DOI]
[28] Vemula SV, Mittal SK. Production of adenovirus vectors and their use as a delivery system for influenza vaccines. Expert Opin Biol Ther, 2010; 10: 1469-1487.[DOI]
[29] Bommareddy PK, Patel A, Hossain S et al. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am J Clin Dermatol, 2017; 18: 1-15.[DOI]
[30] Zainutdinov SS, Kochneva GV, Netesov SV et al. Directed evolution as a tool for the selection of oncolytic RNA viruses with desired phenotypes. Oncolytic Virother, 2019; 8: 9-26.[DOI]
[31] Guo ZS, Lu B, Guo Z et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J Immunother Cancer, 2019; 7: 6.[DOI]
[32] Kim JH, Oh JY, Park BH et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther, 2006; 14: 361-370.[DOI]
[33] Park SH, Breitbach CJ, Lee J et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol Ther, 2015; 23: 1532-1540.[DOI]
[34] Monge BMC, Xie C, Steinberg SM et al. A phase I/II study of Pexa-Vec oncolytic virus in combination with immune checkpoint inhibition in refractory colorectal cancer. J Clin Oncol, 2020; 38: 117-117.[DOI]
[35] Lauer UM, Schell M, Beil J et al. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clin Cancer Res, 2018; 24: 4388-4398.[DOI]
[36] Downs-Canner S, Guo ZS, Ravindranathan R et al. Phase 1 Study of Intravenous Oncolytic Poxvirus (vvDD) in Patients With Advanced Solid Cancers. Mol Ther, 2016; 24: 1492-1501.[DOI]
[37] TBio-6517. Accessed 4 April 2022. Available at:[Web]
[38] Kirn D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: what have we learned? Gene Ther, 2001; 8: 89-98.[DOI]
[40] Philbrick B, Adamson DC. DNX-2401: An investigational drug for the treatment of recurrent glioblastoma. Expert Opin Inv Drug, 2019; 28: 1041-1049.[DOI]
[41] Lang FF, Conrad C, Gomez-Manzano C et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol, 2018; 36: 1419-1427.[DOI]
[42] Chia SL, Lei J, Ferguson DJP et al. Group B adenovirus enadenotucirev infects polarised colorectal cancer cells efficiently from the basolateral surface expected to be encountered during intravenous delivery to treat disseminated cancer. Virology, 2017; 505: 162-171.[DOI]
[43] Eriksson E, Milenova I, Wenthe J et al. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin Cancer Res, 2017; 23: 5846-5857.[DOI]
[44] Wenthe J, Naseri S, Labani-Motlagh A et al. Boosting CAR T-cell responses in lymphoma by simultaneous targeting of CD40/4-1BB using oncolytic viral gene therapy. Cancer Immunol Immun, 2021; 70: 2851-2865.[DOI]
[45] Ranki T, Pesonen S, Hemminki A et al. Phase I study with ONCOS-102 for the treatment of solid tumors - an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer, 2016; 4: 17.[DOI]
[46] Agarwalla PK, Aghi MK. Oncolytic herpes simplex virus engineering and preparation. Methods Mol Biol, 2012; 797: 1-19.[DOI]
[47] Chayavichitsilp P, Buckwalter JV, Krakowski AC et al. Herpes simplex. Pediatr Rev, 2009; 30: 119-130.[DOI]
[48] Carpenter AB, Carpenter AM, Aiken R et al. Oncolytic virus in gliomas: A review of human clinical investigations. Ann Oncol, 2021; 32: 968-982.[DOI]
[49] Geevarghese SK, Geller DA, de Haan HA et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther, 2010; 21: 1119-1128.[DOI]
[50] Dolan A, McKie E, MacLean AR et al. Status of the ICP34.5 gene in herpes simplex virus type 1 strain 17. J Gen Virol, 1992; 73: 971-973.[DOI]
[51] Roizman B. The function of herpes simplex virus genes: a primer for genetic engineering of novel vectors. P Natl Acad Sci Usa, 1996; 93: 11307-11312.[DOI]
[52] Kelly KJ, Wong J, Fong Y. Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin Inv Drug, 2008; 17: 1105-1113.[DOI]
[53] Bernstock JD, Bag AK, Fiveash J et al. Design and Rationale for First-in-Human Phase 1 Immunovirotherapy Clinical Trial of Oncolytic HSV G207 to Treat Malignant Pediatric Cerebellar Brain Tumors. Hum Gene Ther, 2020; 31: 1132-1139.[DOI]
[54] Haines BB, Denslow A, Grzesik P et al. ONCR-177, an Oncolytic HSV-1 Designed to Potently Activate Systemic Antitumor Immunity. Cancer Immunol Res, 2021; 9: 291-308.[DOI]
[55] Zhang B, Huang J, Tang J et al. Intratumoral OH2, an oncolytic herpes simplex virus 2, in patients with advanced solid tumors: A multicenter, phase I/II clinical trial. J Immunother Cancer, 2021; 9.[DOI]
[56] Wang Y, Zhou X, Wu Z et al. Preclinical Safety Evaluation of Oncolytic Herpes Simplex Virus Type 2. Hum Gene Ther, 2019; 30: 651-660.[DOI]
[57] Zawit M, Swami U, Awada H et al. Current status of intralesional agents in treatment of malignant melanoma. Ann Transl Med, 2021; 9: 1038.[DOI]
[58] Tayeb S, Zakay-Rones Z, Panet A. Therapeutic potential of oncolytic Newcastle disease virus: a critical review. Oncolytic Virother, 2015; 4: 49-62.[DOI]
[59] Freeman AI, Zakay-Rones Z, Gomori JM et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther, 2006; 13: 221-228.[DOI]
[60] Csatary LK, Moss RW, Beuth J et al. Beneficial treatment of patients with advanced cancer using a Newcastle disease virus vaccine (MTH-68/H). Anticancer Res, 1999; 19: 635-638.
[61] Keshavarz M, Nejad ASM, Esghaei M et al. Oncolytic Newcastle disease virus reduces growth of cervical cancer cell by inducing apoptosis. Saudi J Biol Sci, 2020; 27: 47-52.[DOI]
[62] Goetz C, Dobrikova E, Shveygert M et al. Oncolytic poliovirus against malignant glioma. Future Virol, 2011; 6: 1045-1058.[DOI]
[63] Takai Y, Miyoshi J, Ikeda W et al. Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat Rev Mol Cell Biol, 2008; 9: 603-615.[DOI]
[64] Desjardins A, Gromeier M, Herndon JE et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med, 2018; 379: 150-161.[DOI]
[65] Beasley GM, Nair SK, Farrow NE et al. Phase I trial of intratumoral PVSRIPO in patients with unresectable, treatment-refractory melanoma. J Immunother Cancer, 2021; 9: e002203.[DOI]
[66] Gromeier M, Nair SK. Recombinant Poliovirus for Cancer Immunotherapy. Annu Rev Med, 2018; 69: 289-299.[DOI]
[67] Merrill MK, Bernhardt G, Sampson JH et al. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro Oncol, 2004; 6: 208-217.[DOI]
[68] Snyder AJ, Wang JC, Danthi P. Components of the Reovirus Capsid Differentially Contribute to Stability. J Virol, 2019; 93: 10-1128.[DOI]
[69] Lelli D, Moreno A, Steyer A et al. Detection and Characterization of a Novel Reassortant Mammalian Orthoreovirus in Bats in Europe. Viruses, 2015; 7: 5844-5854.[DOI]
[70] Maitra R, Ghalib MH, Goel S. Reovirus: A targeted therapeutic--progress and potential. Mol Cancer Res, 2012; 10: 1514-1525.[DOI]
[71] Kicielinski KP, Chiocca EA, Yu JS et al. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther, 2014; 22: 1056-1062.[DOI]
[72] Thompson B. Oncolytics Biotech® Inc.: REOLYSIN® for melanoma therapy. Melanoma Manag, 2015; 2: 105-107.[DOI]
[73] Carew JS, Espitia CM, Zhao W et al. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis, 2013; 4: e728.[DOI]
[74] Jaime-Ramirez AC, Yu JG, Caserta E et al. Reolysin and Histone Deacetylase Inhibition in the Treatment of Head and Neck Squamous Cell Carcinoma. Mol Ther Oncolytics, 2017; 5: 87-96.[DOI]
[75] Donina S, Strele I, Proboka G et al. Adapted ECHO-7 virus Rigvir immunotherapy (oncolytic virotherapy) prolongs survival in melanoma patients after surgical excision of the tumour in a retrospective study. Melanoma Res, 2015; 25: 421-426.[DOI]
[76] Alberts P, Tilgase A, Rasa A et al. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur J Pharmacol, 2018; 837: 117-126.[DOI]
[77] Alberts P, Olmane E, Brokāne L et al. Long-term treatment with the oncolytic ECHO-7 virus Rigvir of a melanoma stage IV M1c patient, a small cell lung cancer stage IIIA patient, and a histiocytic sarcoma stage IV patient-three case reports. Apmis, 2016; 124: 896-904.[DOI]
[78] Tilgase A, Olmane E, Nazarovs J et al. Multimodality Treatment of a Colorectal Cancer Stage IV Patient with FOLFOX-4, Bevacizumab, Rigvir Oncolytic Virus, and Surgery. Case Rep Gastroenterol, 2018; 12: 457-465.[DOI]
[79] Pumpure E, Dručka E, Kigitoviča D et al. Management of a primary malignant melanoma of uterine cervix stage IVA patient with radical surgery and adjuvant oncolytic virus Rigvir® therapy: A case report. Clin Case Rep, 2020; 8: 1538-1543.[DOI]
[80] RIGVIR MARKETING AUTHORISATION SUSPENDED; INFORMATION FOR CURRENT PATIENTS. On 30 May 2019, the State Agency of Medicines (Agency) suspended marketing authorisation of the medicinal product “Rigvir solution for injections” (hereinafter-Rigvir). Accessed 28 Febuary 2022. Available at:[Web]
[81] Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst, 2006; 98: 298-3 00.[DOI]
[82] Dobner T, Horikoshi N, Rubenwolf S et al. Blockage by adenovirus E4orf6 of transcriptional activation by the p53 tumor suppressor. Science, 1996; 272: 1470-1473.[DOI]
[83] Benedict CA, Norris PS, Prigozy TI et al. Three adenovirus E3 proteins cooperate to evade apoptosis by tumor necrosis factor-related apoptosis-inducing ligand receptor-1 and -2. J Biol Chem, 2001; 276: 3270-3278.[DOI]
[84] Lei J, Li QH, Yang JL et al. The antitumor effects of oncolytic adenovirus H101 against lung cancer. Int J Oncol, 2015; 47: 555-562.[DOI]
[85] He CB, Lao XM, Lin XJ. Transarterial chemoembolization combined with recombinant human adenovirus type 5 H101 prolongs overall survival of patients with intermediate to advanced hepatocellular carcinoma: A prognostic nomogram study. Chin J Cancer, 2017; 36: 59.[DOI]
[86] Zhang R, Cui Y, Guan X et al. A Recombinant Human Adenovirus Type 5 (H101) Combined With Chemotherapy for Advanced Gastric Carcinoma: A Retrospective Cohort Study. Front Oncol, 2021; 11: 752504.[DOI]
[87] Liu BL, Robinson M, Han ZQ et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther, 2003; 10: 292-303.[DOI]
[88] Ramelyte E, Tastanova A, Balázs Z et al. Oncolytic virotherapy-mediated anti-tumor response: A single-cell perspective. Cancer Cell, 2021; 39: 394-406.[DOI]
[89] Heinzerling L, Künzi V, Oberholzer PA et al. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood, 2005; 106: 2287-2294.[DOI]
[90] Johnson DB, Puzanov I, Kelley MC. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy, 2015; 7: 611-619.[DOI]
[91] Hercus TR, Thomas D, Guthridge MA et al. The granulocyte-macrophage colony-stimulating factor receptor: Linking its structure to cell signaling and its role in disease. Blood, 2009; 114: 1289-1298.[DOI]
[92] Willemze R, Cerroni L, Kempf W et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood, 2019; 133: 1703-1714.[DOI]
[93] Ressler JM, Karasek M, Koch L et al. Real-life use of talimogene laherparepvec (T-VEC) in melanoma patients in centers in Austria, Switzerland and Germany. J Immunother Cancer, 2021; 9.[DOI]
[94] Puzanov I, Milhem MM, Minor D et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J Clin Oncol, 2016; 34: 2619-2626.[DOI]
[95] Harrington KJ, Andtbacka RH, Collichio F et al. Efficacy and safety of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in patients with stage IIIB/C and IVM1a melanoma: subanalysis of the Phase III OPTiM trial. Onco Targets Ther, 2016; 9: 7081-7093.[DOI]
[96] Todo T, Martuza RL, Rabkin SD et al. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A, 2001; 98: 6396-6401.[DOI]
[97] Cassady KA, Gross M, Roizman B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol, 1998; 72: 7005-7011.[DOI]
[98] York IA, Roop C, Andrews DW et al. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell, 1994; 77: 525-535.[DOI]
[99] Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol, 1988; 62: 196-205.[DOI]
[100] Uchihashi T, Nakahara H, Fukuhara H et al. Oncolytic herpes virus G47Δ injected into tongue cancer swiftly traffics in lymphatics and suppresses metastasis. Mol Ther Oncolytics, 2021; 22: 388-398.[DOI]
[101] Raja J, Ludwig JM, Gettinger SN et al. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer, 2018; 6: 140.[DOI]
[102] Hong J, Yun CO. Overcoming the limitations of locally administered oncolytic virotherapy. BMC Biomed Eng, 2019; 1: 17.[DOI]
[103] Murin CD, Wilson IA, Ward AB. Antibody responses to viral infections: A structural perspective across three different enveloped viruses. Nat Microbiol, 2019; 4: 734-747.[DOI]
[104] Ye X, Xiao L, Zheng X et al. Seroprevalence of Neutralizing Antibodies to Human Adenovirus Type 4 and 7 in Healthy Populations From Southern China. Front Microbiol, 2018; 9: 3040.[DOI]
[105] Muñoz-Alía MÁ, Nace RA, Tischer A et al. MeV-Stealth: A CD46-specific oncolytic measles virus resistant to neutralization by measles-immune human serum. PLoS Pathog, 2021; 17: e1009283.[DOI]
[106] Smith JG, Cassany A, Gerace L et al. Nemerow, Neutralizing antibody blocks adenovirus infection by arresting microtubule-dependent cytoplasmic transport. J Virol, 2008; 82: 6492-6500.[DOI]
[107] Shin DH, Nguyen T, Ozpolat B et al. Current strategies to circumvent the antiviral immunity to optimize cancer virotherapy. J Immunother Cancer, 2021; 9.[DOI]
[108] Ying L, Cheng H, Xiong XW et al. Interferon alpha antagonizes the anti-hepatoma activity of the oncolytic virus M1 by stimulating anti-viral immunity. Oncotarget, 2017; 8: 24694-24705.[DOI]
[109] Sarén T, Ramachandran M, Martikainen M et al. Insertion of the Type-I IFN Decoy Receptor B18R in a miRNA-Tagged Semliki Forest Virus Improves Oncolytic Capacity but Results in Neurotoxicity. Mol Ther Oncolytics, 2017; 7: 67-75.[DOI]
[110] Xia M, Luo D, Dong J et al. Graphene oxide arms oncolytic measles virus for improved effectiveness of cancer therapy. J Exp Clin Cancer Res, 2019; 38: 408.[DOI]
[111] Wang Y, Huang H, Zou H et al. Liposome Encapsulation of Oncolytic Virus M1 To Reduce Immunogenicity and Immune Clearance in Vivo. Mol Pharm, 2019; 16: 779-785.[DOI]
[112] Lv P, Liu X, Chen X et al. Genetically Engineered Cell Membrane Nanovesicles for Oncolytic Adenovirus Delivery: A Versatile Platform for Cancer Virotherapy. Nano Lett, 2019; 19: 2993-3001.[DOI]
[113] Ozcan G, Ozpolat B, Coleman RL et al. Preclinical and clinical development of siRNA-based therapeutics. Adv Drug Deliv Rev, 2015; 87: 108-119.[DOI]
[114] Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol, 2018; 15. 366-381.[DOI]
[115] Everts A, Bergeman M, McFadden G et al. Simultaneous Tumor and Stroma Targeting by Oncolytic Viruses. Biomedicines, 2020; 8.[DOI]
[116] Zheng M, Huang J, Tong A et al. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol Ther Oncolytics, 2019; 15: 234-247.[DOI]
[117] Vähä-Koskela M, Hinkkanen A. Tumor restrictions to oncolytic virus. Biomedicines, 2014; 2: 163-194.[DOI]
[118] Santi A, Kugeratski FG, Zanivan S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics, 2018; 18: e1700167.[DOI]
[119] Henke E, Nandigama R, Ergün S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci, 2019; 6: 160.[DOI]
[120] Choi H, Moon A. Crosstalk between cancer cells and endothelial cells: implications for tumor progression and intervention. Arch Pharm Res, 2018; 41: 711-724.[DOI]
[121] Maishi N, Hida K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci, 2017; 108: 1921-1926.[DOI]
[122] Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol, 2019; 10: 1835.[DOI]
[123] Liu Z, Kuang W, Zhou Q et al. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma?cells via the SMAD2/3 signalling pathway. Int J Mol Med, 2018; 42: 3395-3403.[DOI]
[124] Sette P, Amankulor N, Li A et al. GBM-Targeted oHSV Armed with Matrix Metalloproteinase 9 Enhances Anti-tumor Activity and Animal Survival. Mol Ther Oncolytics, 2019; 15: 214-222.[DOI]
[125] Fingleton B. Matrix metalloproteinases: Roles in cancer and metastasis. Front Biosci, 2006; 11: 479-491.[DOI]
[126] Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev Immunol, 2012; 32: 463-488.[DOI]
[127] Kulkarni A, Chandrasekar V, Natarajan SK et al. A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat Biomed Eng, 2018; 2: 589-599.[DOI]
[128] Zheng X, Turkowski K, Mora J et al. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget, 2017; 8: 48436-48452.[DOI]
[129] Rao L, Zhao SK, Wen C et al. Activating Macrophage-Mediated Cancer Immunotherapy by Genetically Edited Nanoparticles. Adv Mater, 2020; 32: e2004853.[DOI]
[130] Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol, 2018; 18: 153-167.[DOI]
[131] Herbst RS, Soria JC, Kowanetz M et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature, 2014; 515: 563-567.[DOI]
[132] Tumeh PC, Harview CL, Yearley JH et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 2014; 515: 568-571.[DOI]
[133] Miggelbrink AM, Jackson JD, Lorrey SJ et al. CD4 T-Cell Exhaustion: Does It Exist and What Are Its Roles in Cancer? Clin Cancer Res, 2021; 27: 5742-5752.[DOI]
[134] Ribas A, Chesney J, Long GV et al. 1037O MASTERKEY-265: A phase III, randomized, placebo (Pbo)-controlled study of talimogene laherparepvec (T) plus pembrolizumab (P) for unresectable stage IIIB–IVM1c melanoma (MEL). Ann Oncol, 2021; 32: S868-S869.[DOI]
[135] Saito Y, Sunamura M, Motoi F et al. Oncolytic replication-competent adenovirus suppresses tumor angiogenesis through preserved E1A region. Cancer Gene Ther, 2006; 13: 242-252.[DOI]
[136] Motoi F, Sunamura M, Ding L et al. Effective gene therapy for pancreatic cancer by cytokines mediated by restricted replication-competent adenovirus. Hum Gene Ther, 2000; 11: 223-235.[DOI]
[137] Seymour LW, Fisher KD. Oncolytic viruses: Finally delivering. Br J Cancer, 2016; 114: 357-361.[DOI]
[138] Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol, 2012; 30: 658-670.[DOI]
[139] Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662.[DOI]
[140] Russell S, Miller A. Heterogeneous delivery is a barrier to the translational advancement of oncolytic virotherapy for treating solid tumors. Virus Adapt Treat, 2014; 6: 11-31.[DOI]
[141] Allan KJ, Stojdl DF, Swift SL. High-throughput screening to enhance oncolytic virus immunotherapy. Oncolytic Virother, 2016; 5: 15-25.[DOI]
[142] Bailey K, Kirk A, Naik S et al. Mathematical model for radial expansion and conflation of intratumoral infectious centers predicts curative oncolytic virotherapy parameters. PLoS One, 2013; 8: e73759.[DOI]
[143] Yokoda R, Nagalo BM, Vernon B et al. Oncolytic virus delivery: from nano-pharmacodynamics to enhanced oncolytic effect. Oncolytic Virother, 2017; 6: 39-49.[DOI]
[144] Seyed-Khorrami SM, Soleimanjahi H, Soudi S et al. MSCs loaded with oncolytic reovirus: migration and in vivo virus delivery potential for evaluating anti-cancer effect in tumor-bearing C57BL/6 mice. Cancer Cell Int, 2021; 21: 244.[DOI]
[145] Ghaleh HEG, Vakilzadeh G, Zahiri A et al. Investigating the potential of oncolytic viruses for cancer treatment via MSC delivery. Cell Commun Signal, 2023; 21: 228.[DOI]


