A Beginner's Guide to Genomics in Complex Neurological Disorders
Trudy Quirke1, Roy D. Sleator1*
1Department of Biological Sciences, Munster Technological University, Cork, Ireland
*Correspondence to: Roy D. Sleator, PhD, Professor, Department of Biological Sciences, Munster Technological University, Bishopstown, Cork, T12 P928, Ireland; Email: roy.sleator@mtu.ie
DOI: 10.53964/id.2024029
Abstract
In view of the recent progress in genomics and next-generation sequencing technologies, we consider here their contribution toward neurological disorders. We describe the revolutionary impact that whole-genome and whole-exome sequencing have had on diagnostics and the identification of important genetic loci associated with a range of disorders. Neurological disorders affecting the central and peripheral nervous systems are complex, both in diagnosis and treatment. Herein we show that by applying multiomic tools to assist researchers in identifying new biomarkers and therapeutic approaches, the diagnosis, treatment, and prognosis of these disorders can be significantly improved.
Keywords: neurological disorders, genomics, next-generation sequencing, whole-genome sequencing, whole-exome sequencing, multi-omics, systems biology
1 INTRODUCTION
Neurodegenerative illnesses, neurodevelopmental disorders, neuroinflammatory ailments, and a variety of neuropsychiatric disorders can be generally incorporated into the broad category of neurological disorders[1]. This range includes common disorders such as Alzheimerʼs disease (AD)[2] and Parkinson’s disease (PD)[3], as well as less common inherited illnesses including familial hemiplegic migraine[4] and Huntingtonʼs disease[5], among others[6]. These disorders are characterized by numerous and diverse clinical symptoms, and complex pathophysiological mechanisms, which have far-reaching implications for the patient’s health and wellbeing[7]. For example, neurodegenerative disorders such as Huntingtonʼs disease and AD are characterised by protein aggregation and neuronal loss, while neuroinflammatory disorders like multiple sclerosis (MS) involve immune-mediated damage to the nervous system[8]. Additionally, neurodegeneration results from the mutant huntingtin protein aggregating within neurones in Huntingtonʼs disease, which impairs transcription and mitochondrial function[9].
While there are many rare neurological disorders whose precise aetiology is still unclear[10], significant recent advances have been made in biomarker discovery. One such rare neurological disorder is familial hemiplegic migraine type 2 (FHM2), which is linked to mutations in the ATP1A2 gene. This mutation alters the extracellular potassium and glutamate clearance process by impairing the activity of the Na+/K+ ATPase in astrocytes. Comprehension of the complicated symptomatology of FHM2, which includes excruciating headaches, hemiplegia, and, in certain cases, seizures and cognitive impairment, requires a comprehension of this pathophysiological process[11]. Another example is Rett syndrome, caused by mutations in the gene MECP2. Mutations in this gene disrupt redox regulation pathways, leading to mitochondrial dysfunction and resulting in severe neurological and systemic symptoms in patients. It underpins once more the complication of the disorder due to variability in the clinical manifestations, including loss of motor skills and severe cognitive impairments of the patients according to the nature of the mutations in MECP2[12]. These disorders, discussed in further sections, have made advancements possible by using a systems biology approach facilitated by the development of omics technologies[13].
The term “omics” denotes the interdisciplinary exploration and examination of four primary domains[14]: genomics (exploration of genes), transcriptomics (investigation of gene expression), proteomics (study of proteins), and metabolomics (analysis of metabolites). Omics technologies have also expanded to include the study of metagenomics (microbial diversity), epigenomics (changes caused by modification of gene expression), lipidomics (lipid profiles) and microbiomics (microbiota), amongst others[15,16].
In the interest of brevity, we will focus only on recent clinical applications of genomics, with a case study lead analysis of how next generation sequencing technologies (specifically whole-genome sequencing (WGS), whole-exome sequencing (WES), targeted sequencing (TS), and genome wide association studies (GWAS), has improved our understanding of key neurological disorders (Figure 1). Genomic analysis not only provides valuable insights into disease mechanisms[17], it also aids in the development of personalised medicine, genetic testing, and risk assessment by identifying the genes linked to these disorders[18].
|
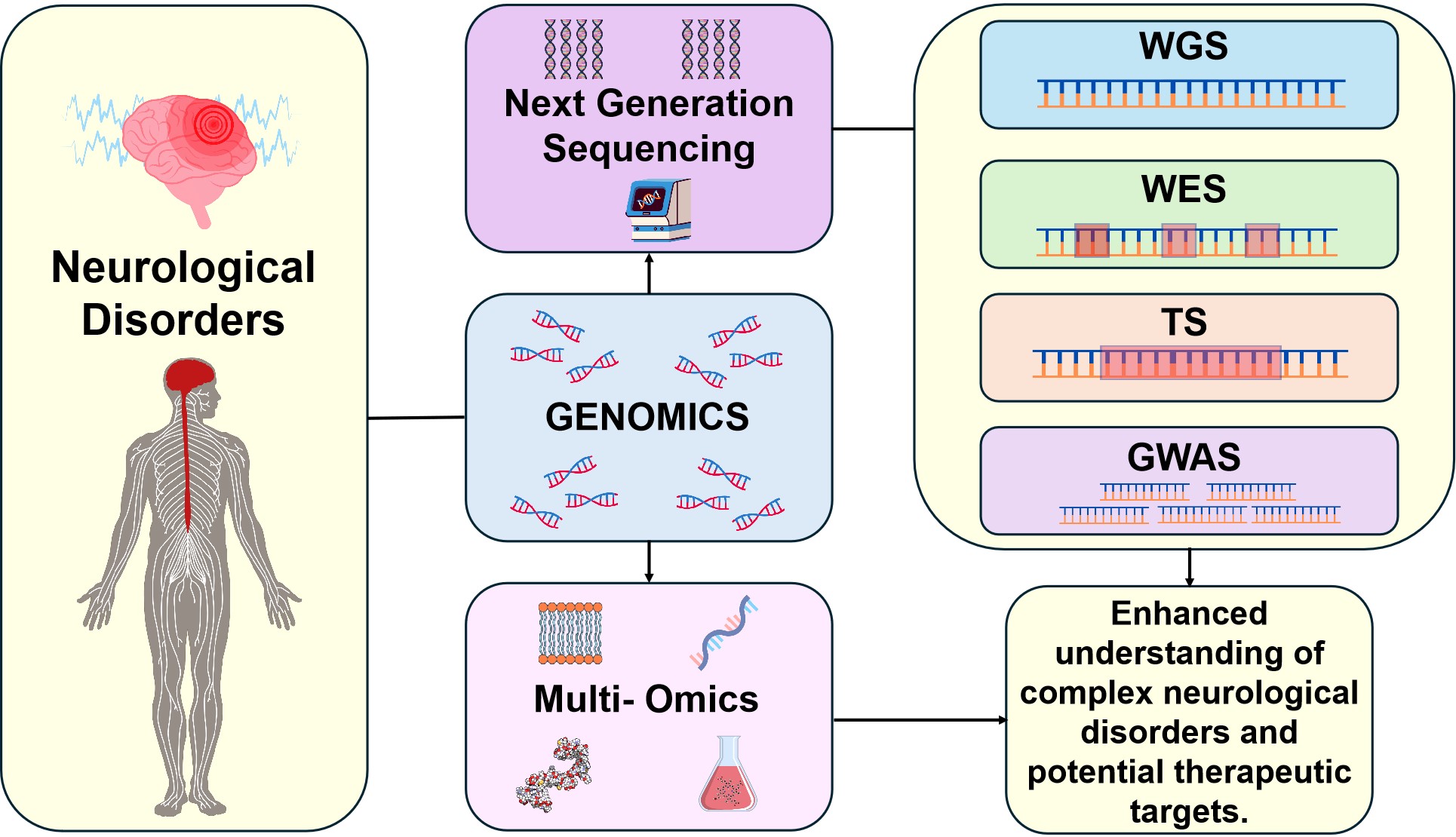
Figure 1. Schematic Representation of the Role of Next-Generation Sequencing (NGS) Technologies and Genomic Approaches in Enhancing the Understanding of Complex Neurological Disorders.
As for Figure 1, it how genomic techniques, including WGS, WES, TS, and GWAS, contribute to identifying disease mechanisms and potential therapeutic targets in various neurological conditions. Additionally, it highlights the use of genomics with other omic-technologies to help better understand the intricacies of neurological disorders.
2 NEUROLOGICAL DISORDERS
The more complex the neurological disorder, the more important it becomes to trace how these diseases manifest within the various physiological systems. In aiming to understand the relationship between neurological disorders and their clinical presentation, more effective diagnostic and treatment strategies can be devised. While neurological disorders principally impact the nervous system, their consequences frequently transcend these bounds[19]. Beyond psychological implications, secondary impacts can infiltrate diverse physiological domains (Figure 2). The muscular and sensory systems may experience altered function, while the endocrine, cardiovascular and respiratory apparatus can be additionally impacted. More remote regions such as the gastrointestinal, urinary and immune systems can also fall subject to the indirect implications of certain critical neurological conditions[20]. The spread and seriousness of these supplementary impacts are contingent on the character and gravity of the underlying nervous system issue. For example, PD can affect the autonomic nervous system, gastrointestinal, sensory, musculoskeletal and cardiovascular systems[21-23].
|
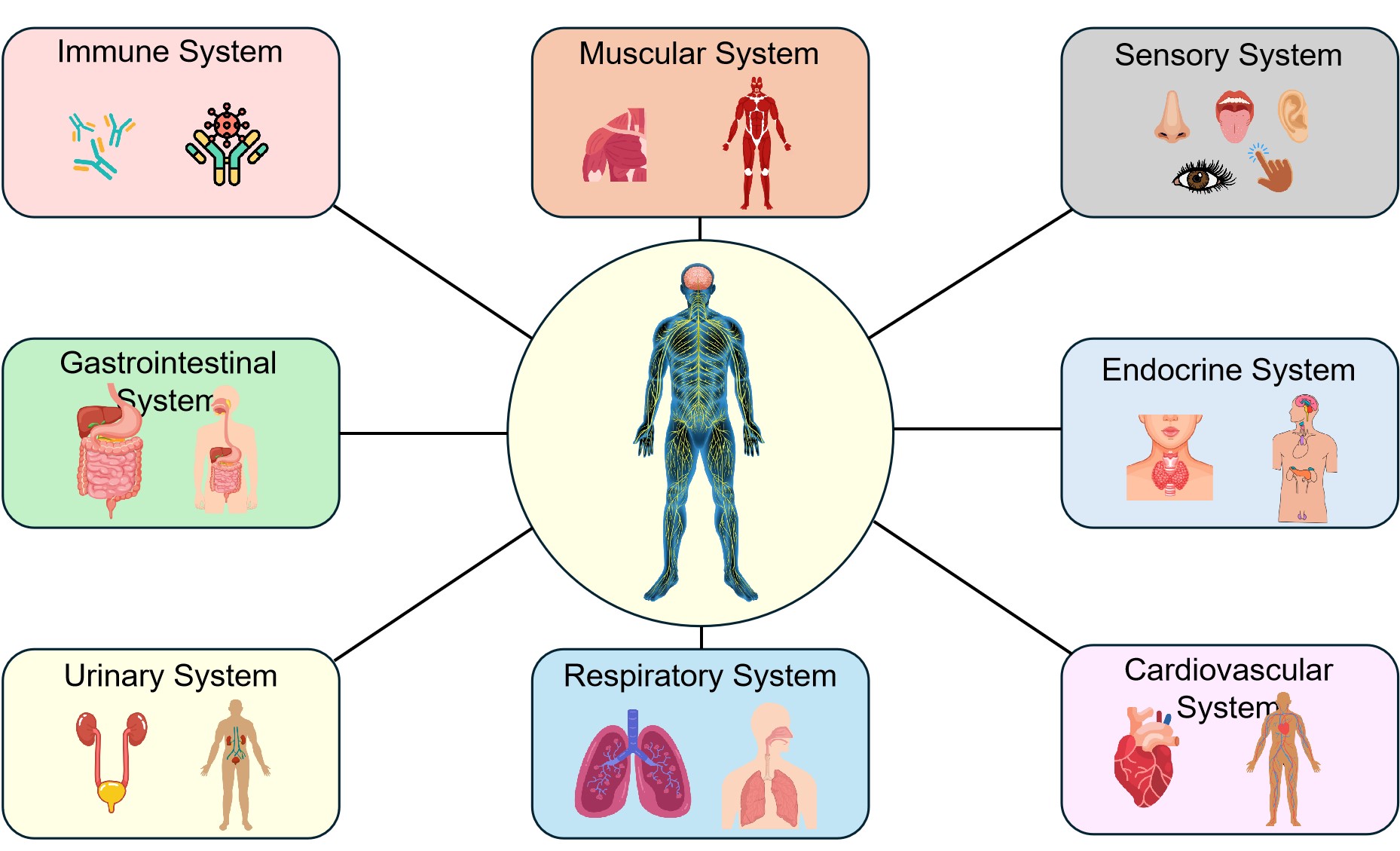
Figure 2. The Wide-Ranging Effects of Neurological Disorders Beyond the Central Nervous System, Highlighting How Conditions Like PD Can Impact the Muscular, Sensory, Cardiovascular, and Gastrointestinal Systems, Among Others. The illustration shows that the severity and nature of the neurological disorder determines the extent of these effects, which can also spread to the endocrine, respiratory, urinary, and immune systems.
Ataxias, encephalopathies, genetic forms of brain abnormalities, myopathies and muscular dystrophies, neuropathies, and different types of dementia represent just a few of the many clinical manifestations of neurological disorders[24]. Furthermore, there is a considerable phenotypic overlap among several neurological illness types, making accurate diagnosis often challenging[25] (Figure 3). Hemiplegic migraine and transient ischemic attack, for example, both exhibit similar reversibility of weakness on one side of the body, as well as dizziness and speech problems, leading to difficulty in diagnosis, as reported by Shao et al[26]. This complexity means that genetic testing of the patient, together with family history, are required to ensure accurate diagnosis, and appropriate intervention.
|
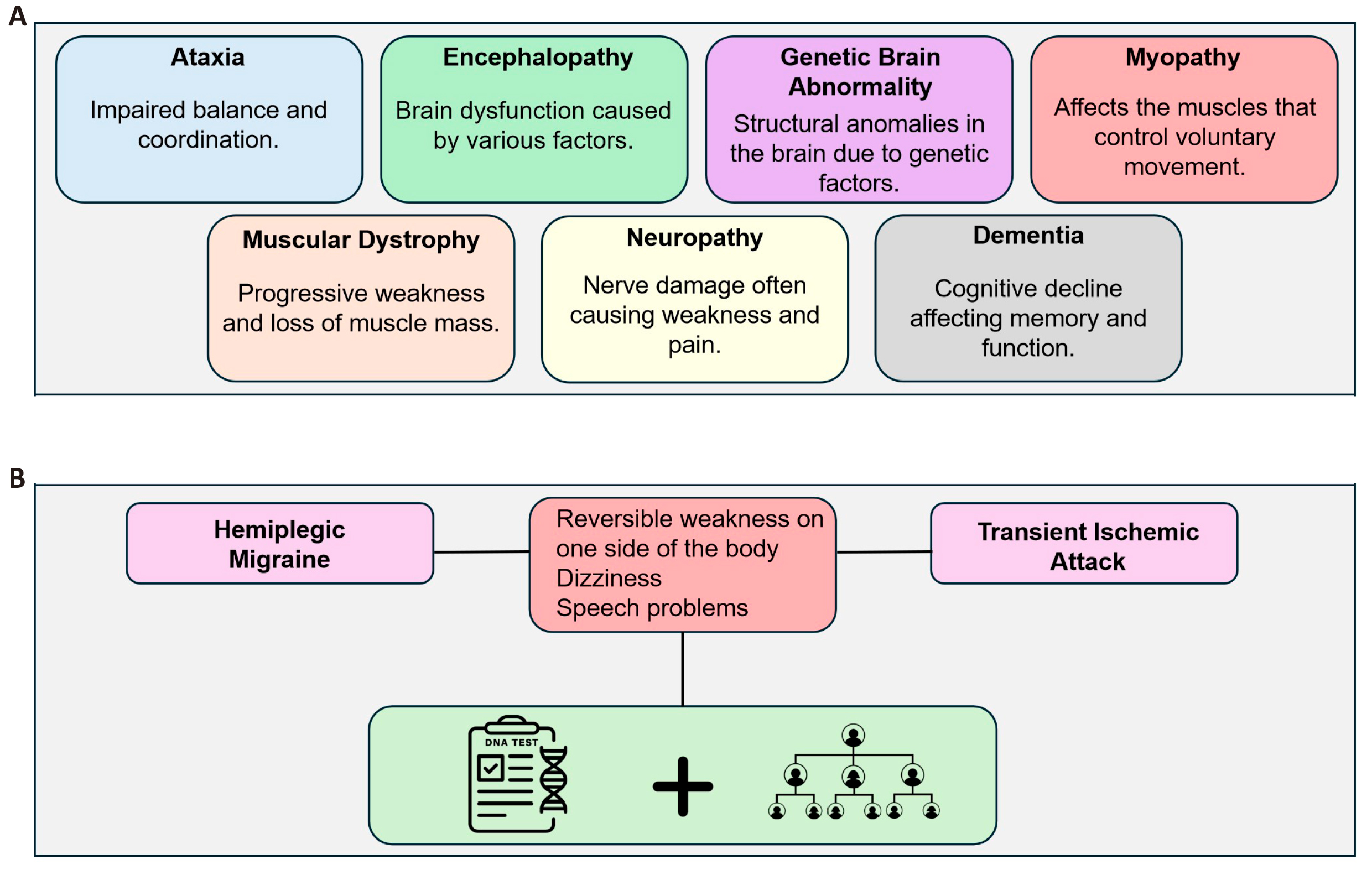
Figure 3. The Clinical Manifestations and Diagnostic Challenges for a Variety of Neurological Disorders. Panel A shows the symptomatology of different conditions, while Panel B shows how hard the differential diagnosis can be between conditions like hemiplegic migraine and transient ischemic attack due to overlapping symptoms. The figure further helps to reiterate the part genetic testing and family history play in arriving at an accurate diagnosis, followed by appropriately targeted treatment.
Genomics, and more specifically genomic sequencing, has enabled the identification of key genetic loci linked to neurological disorders[27]. Such genomic interrogation has revolutionised the study of neurological disorders in recent years[28], enabling the development of improved diagnostic tests[29], as well as improved prognostic outcomes[30].
In summary, most neurological disorders present as a very wide and often overlapping spectrum of clinical symptoms, which makes them very hard to diagnose precisely. Because of this, advanced diagnostic techniques like genome sequencing are required to find underlying genetic variables and enhance patient outcomes. Herein, we will we provide a brief overview of the key technological developments in the field of DNA sequencing, focusing on some of the more common sequencing technologies applied in the field of complex neurological disorders.
2.1 DNA Sequencing
Traditional diagnostic techniques sometimes fall short of completely appreciating the spectrum of neurological disorders due to their complexity. DNA sequencing can help us understand these conditions by identifying the genetic components that underlie individual experiences.
2.1.1 First Generation Sequencing
The first generation of DNA sequencing, developed by Frederick Sanger in 1977, uses chain-terminating inhibitors to sequence DNA. This method, known for its high accuracy, involves the incorporation of dideoxynucleotides that terminate DNA strand elongation, allowing for the determination of the DNA sequence. Despite its accuracy, Sanger sequencing is time-consuming and labour-intensive, making it less suitable for large-scale projects like whole genome sequencing[31,32]. Techniques like the Sanger “plus and minus” method and the Maxam-Gilbert chemical cleavage method relied heavily on electrophoresis and radiolabelling, which were complex and onerous[32,33]. Improvements in Sanger sequencing, including the use of fluorescence-based detection and capillary electrophoresis, led to the development of automated DNA sequencing machines, significantly enhancing sequencing speed and accuracy[31,33]. For a summary of the first-generation sequencing technologies, including their respective strengths, weaknesses and applications, we refer the reader to Table 1.
Table 1. Summery of First Generation Sequencing
Advantages |
Disadvantages |
Applications |
||
First gen. |
Sanger |
High accuracy; Robust; Widely adopted; Suitable for small-scale sequencing projects; Well-understood technology. |
Limited read length; Labour intensive; Low throughput; High cost per base sequenced compared to newer technologies. |
Highly accurate sequencing of small DNA fragments, often used for confirming mutations identified by other methods. Itʼs useful in clinical diagnostics for specific, small-scale sequencing tasks, such as validating variants detected by NGS |
Maxam-Gilbert |
No need for cloning; Effective for small nucleotide polymers. |
Complex; Hazardous materials; Not suitable for large-scale sequencing projects; Largely obsolete |
||
Notes: This table summarizes the key features, advantages, disadvantages, and applications of first-generation sequencing techniques, including Sanger and Maxam - Gilbert methods. It highlights the accuracy and robustness of these techniques while noting their limitations in scalability and complexity.
2.1.2 Second Generation Sequencing
Second Generation sequencing methods involve the simultaneous sequencing of millions of small DNA fragments, significantly increasing speed and reducing costs compared to Sanger sequencing[31,32]. Pyrosequencing, developed by Pål Nyrén and commercialized by 454 Life Sciences, was a key innovation, allowing for real-time sequencing without the need for gel electrophoresis. However, this method struggled with homopolymer sequences[33]. The most impactful second-generation technology was Illumina’s sequencing by synthesis (SBS), which used fluorescent reversible terminators and bridge amplification to achieve high accuracy and throughput. Other notable second generation methods included SOLiD sequencing, which used ligation instead of synthesis, and Ion Torrent sequencing, which measured pH changes instead of fluorescence, though each had its limitations in terms of read length and homopolymer detection[33]. For a summary of the second-generation sequencing technologies, including their respective strengths, weaknesses and applications, we refer the reader to Table 2.
Table 2. Summery of Second Generation Sequencing
Sequencing |
Advantages |
Disadvantages |
Applications |
|
Second gen. |
Roche 454 Pyrosequencing |
Faster than sanger. More scalable. High-throughput. Real-time bioluminetric detection. Longer reads. |
Difficulty in accurately reading homopolymer sequences. Higher cost per base compared to other NGS technologies. Technology is becoming obsolete. |
WGS: Offers comprehensive analysis of the entire genome, crucial for identifying genetic variations across coding and non-coding regions. Itʼs particularly useful in diagnosing rare diseases and in research focused on complex genetic disorders like Parkinson’s disease.
WES: Focuses on protein-coding regions of the genome and is widely used in clinical diagnostics, especially for identifying disease-causing mutations in genes related to neurological disorders.
TS: Targets specific genes or genomic regions, making it cost-effective and efficient for clinical diagnostics. It’s particularly beneficial for diagnosing known genetic conditions and for research on specific diseases. GWAS: Identifies genetic markers associated with traits or disease risk across populations. GWAS has been instrumental in understanding the genetic basis of complex diseases like Alzheimer’s and Parkinson’s disease. |
Illumina |
High accuracy. High-throughput. Suitable for a range of applications (WGS, TS & RNA-seq). Paired end reads. Good for short reads. Continuously evolving. |
Shorter read lengths compared to other technologies. Requires precise sample loading to avoid cluster overlap. Initial setup cost can be high. |
||
SOLiD |
Cost effective. High- throughput. High accuracy. |
Short reads. Long run times. Complex data analysis. |
||
Ion Torrent |
Fast. Scalable. Cost effective. Semi-conductor-based detection. |
Problems reading homopolymer regions. Only moderate throughput. Variable read lengths but generally shorter than some other NGS technologies. |
||
Notes: This table provides an overview of second-generation sequencing technologies, such as Roche 454, Illumina, SOLiD, and Ion Torrent. It compares their advantages, including higher throughput and faster sequencing, with their challenges, such as shorter read lengths and difficulty in reading homopolymer sequences.
2.1.3 Third Generation Sequencing
Third-generation sequencing technologies, such as those developed by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), sequence single molecules of DNA in real time. These methods can produce much longer read lengths than next-generation sequencing (NGS), which is beneficial for sequencing repetitive regions of the genome[32,34,35]. Helicos BioSciences pioneered this approach, but it was PacBioʼs Single Molecule Real-Time (SMRT) sequencing that gained widespread use. SMRT sequencing offers real-time monitoring of DNA polymerisation, producing long reads useful for de novo genome assemblies and detecting modified bases[31,33]. The most anticipated development in this generation is nanopore sequencing by ONT, which allows for ultra-long reads and real-time analysis in a compact, portable device. Despite current issues with read accuracy, nanopore sequencing promises significant advancements in speed, cost, and field applicability[31,33,34]. For a summary of the third-generation sequencing technologies, including their respective strengths, weaknesses and applications, we refer the reader to Table 3.
Table 3. Summery of Third Generation Sequencing
Sequencing |
Advantages |
Disadvantages |
Applications |
|
Third gen. |
PacBio SMRT |
Very long read lengths. Real-time sequencing. Can resolve complex genomic regions and repetitive sequences. |
High error rates. Higher cost per base. Significant computational resources needed for data analysis. |
SMRT: Ideal for sequencing repetitive regions and for de novo genome assemblies. Additionally, it helps decipher complicated genomic areas that are difficult for second-generation sequencing technology to grasp and discover structural variations. ONT: Applicable in field-based studies and for rapid sequencing needs. |
Oxford Nanopore |
Ultra long reads. Portable device. Rapid sequencing. |
Lower accuracy. Higher error rates. Requires frequent updates to software and protocols. Lower throughput compared to large-scale Illumina systems. |
||
Notes: This table compares third-generation sequencing technologies, such as PacBio, SMRT, and Oxford Nanopore, highlighting their long-read capabilities and real-time sequencing advantages. It also addresses the challenges, including higher error rates and the need for significant computational resources.
2.1.4 General Workflow of NGS
A typical NGS workflow in a clinical setting (outlined in Figure 4) begins with the procurement of biological samples like blood or tissue from which DNA or RNA is isolated through chemical and mechanical processes[36]. After extraction, DNA or RNA is quantified and checked for quality using spectrophotometry or fluorometry to ensure they are fit for sequencing[37].
|
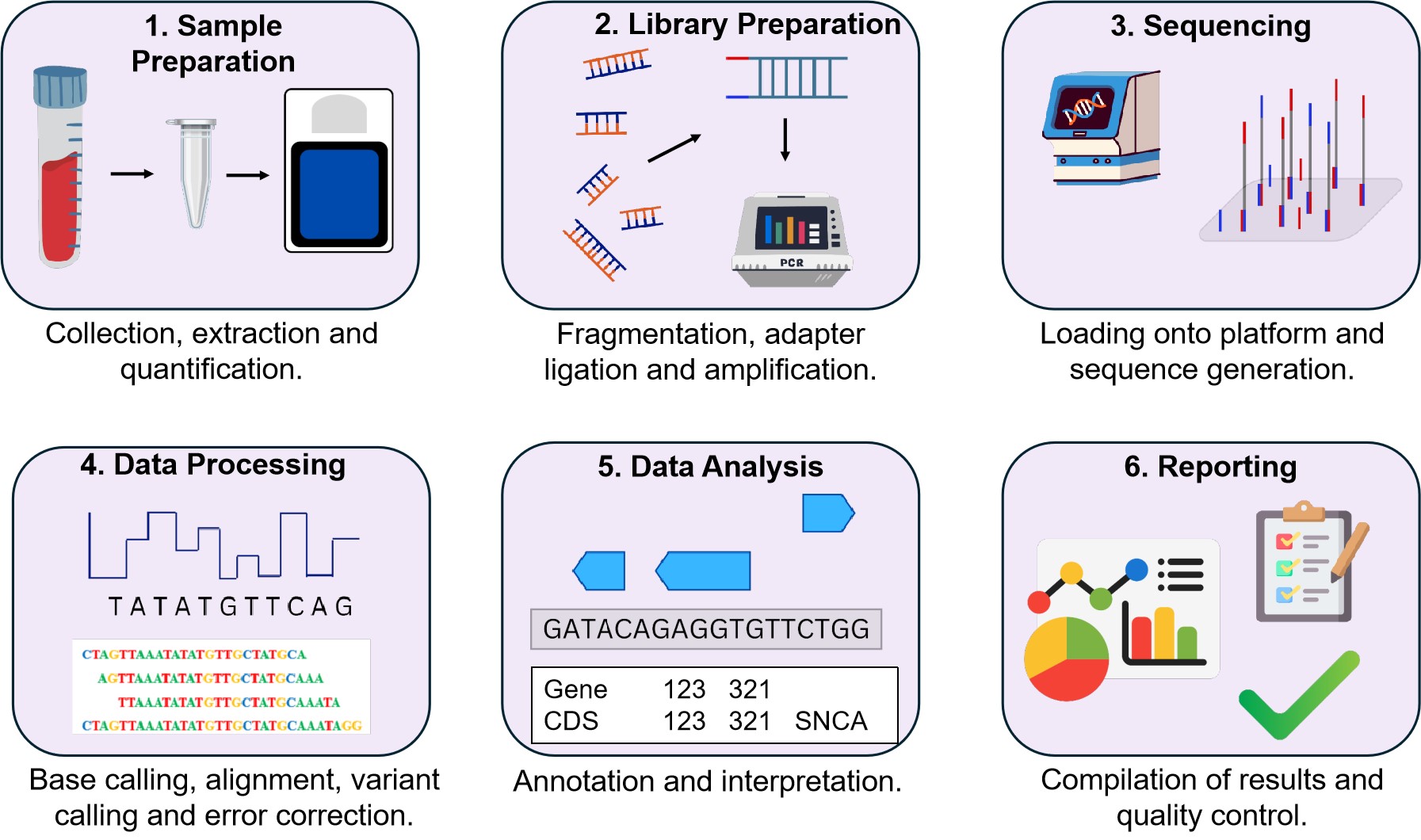
Figure 4. The Workflow of NGS in a Clinical Setting. It details the process from sample collection and DNA/RNA extraction through to sequencing, data processing, and quality control. Each step is crucial for accurate identification of genetic variants and their subsequent clinical interpretation. This overview of NGS workflow involves: 1. Collecting and extracting DNA/RNA from samples, followed by quantification and quality checks. 2. The DNA/RNA is fragmented, adapters are added, and fragments are amplified. 3.Prepared libraries are loaded onto sequencing platforms which produces raw data. 4. & 5. This is then processed to assign bases (A, T, C, G), align to reference genomes, and identify variants, along with annotation. 6. Finally, reports of the accumulated data are created and undergo quality control checks.
During the sample preparation and purification step, the DNA or RNA is sheared into smaller pieces and some short sequences, known as adapters, are annealed to these fragments to allow them to adhere to the sequencing platform and facilitate the subsequent emulsion polymerase chain reaction (PCR), in-situ polonies, or bridge PCR[37,38]. The prepared library is then placed in a sequencer platform[36]. The next stage deals with the sequencing and imaging of DNA fragments. The sequences are synthesised, and their corresponding signals are detected by measuring changes in fluorescence intensity or pH levels, which indicate the incorporation of specific nucleotides during the sequencing process. In Illumina sequencing by synthesis, the fluorescently labelled nucleotides are used for the synthesis process in each cycle and a camera captures images of the number of incorporated nucleotides. This process is repeated several times, thus enabling the sequencer to read the DNA sequence in parts[38].
These sequences are then analysed using programs and techniques like the Basic Local Alignment Search Tool (BLAST), Burrows-Wheeler Aligner (BWA) and Bowtie[37]. BLAST locates areas of local similarity by comparing a query sequence to a database of sequences, enabling the determination of evolutionary and functional links between homologous sequences[39-41]. Another popular software package for aligning DNA sequences to a large reference genome, like the human genome, is BWA. In the fields of computational biology and bioinformatics, BWA is notably well-liked for handling NGS data[42-44]. Additionally, short DNA sequences can be mapped to reference genomes using Bowtie, a quick and memory-efficient aligner that uses a Burrows-Wheeler index to reduce memory utilisation. Because of its ability to align more than 25 million reads per hour, it is an excellent choice for effectively managing large-scale sequencing data[45-47]. Alignment involves matching the sequenced DNA fragments to their corresponding locations on a reference genome map. Once aligned, variant calling is performed to identify differences between the sequenced DNA and the reference genome. This process detects various types of genetic variations, including single nucleotide variants (SNVs), insertions, deletions, and structural variations. Tools such as Genome Analysis Toolkit (GATK) or SAMtools are commonly used to perform these analyses[36-38]. GATK is a collection of command-line tools designed for analysing high-throughput sequencing data with a primary focus on variant discovery[48], while SAMtools is a suite of programs used for interacting with and manipulating high-throughput sequencing data[49]. An example of variant calling can be seen in a study by Skoczylas et al.[50], where TS enabled the identification ofpathogenic variants in genes such as KCNQ and SYNGAP1, which are linked to intellectual disabilities and epilepsy. These findings highlight how variant calling can pinpoint specific genetic mutations that contribute to complex neurological conditions. This study will be discussed in more detail in a later section.
Bioinformatics tools filter out the errors in the sequencing data to ensure high-quality data is obtained. Preprocessing of the raw data and mapping on reference genomes is followed by further bioinformatic analysis[32]. These data are then analysed and annotated to identify the genes and proteins that may be affected by the identified variants and to interpret the biological relevance of these variants with respect to the study or the clinical question under consideration[37]. These interpreted data are then summarised and presented in the form of a report containing information about variants that have been discovered, possible implications of these variants for the patient’s clinical condition, and a suggested course of action or management. Some of the checks put in place for quality controls include sequencing depth, and coverage, as well as error rates[36]. This intricate cycle helps NGS deliver correct genetic information for purposes ranging from experimentation to diagnostics[36-38].
Evolution of technology has brought greater capabilities to the analysis of genetic data, and DNA sequencing has advanced significantly. This is changing the way we identify and treat complicated neurological disorders, and this becomes evident when we talk about NGS. We will examine some practical uses of NGS and its impact on clinical practice in the next section.
3 NGS APPLICATIONS
With the basics of DNA sequencing covered, we now look at how NGS has taken things to the next level. These advanced technological methods not only accelerate the process but also open a wide range of possibilities for clinical applications.
NGS technologies have significantly improved genomic accessibility for clinical applications by making sequencing quicker, more precise, and less expensive than traditional approaches[51,52]. WGS[53], WES[54] and TS[55] (Figure 5) have, in recent years, gained widespread application and popularity in the clinic, facilitating the discovery of key genetic risk factors and disease biomarkers[56].
|
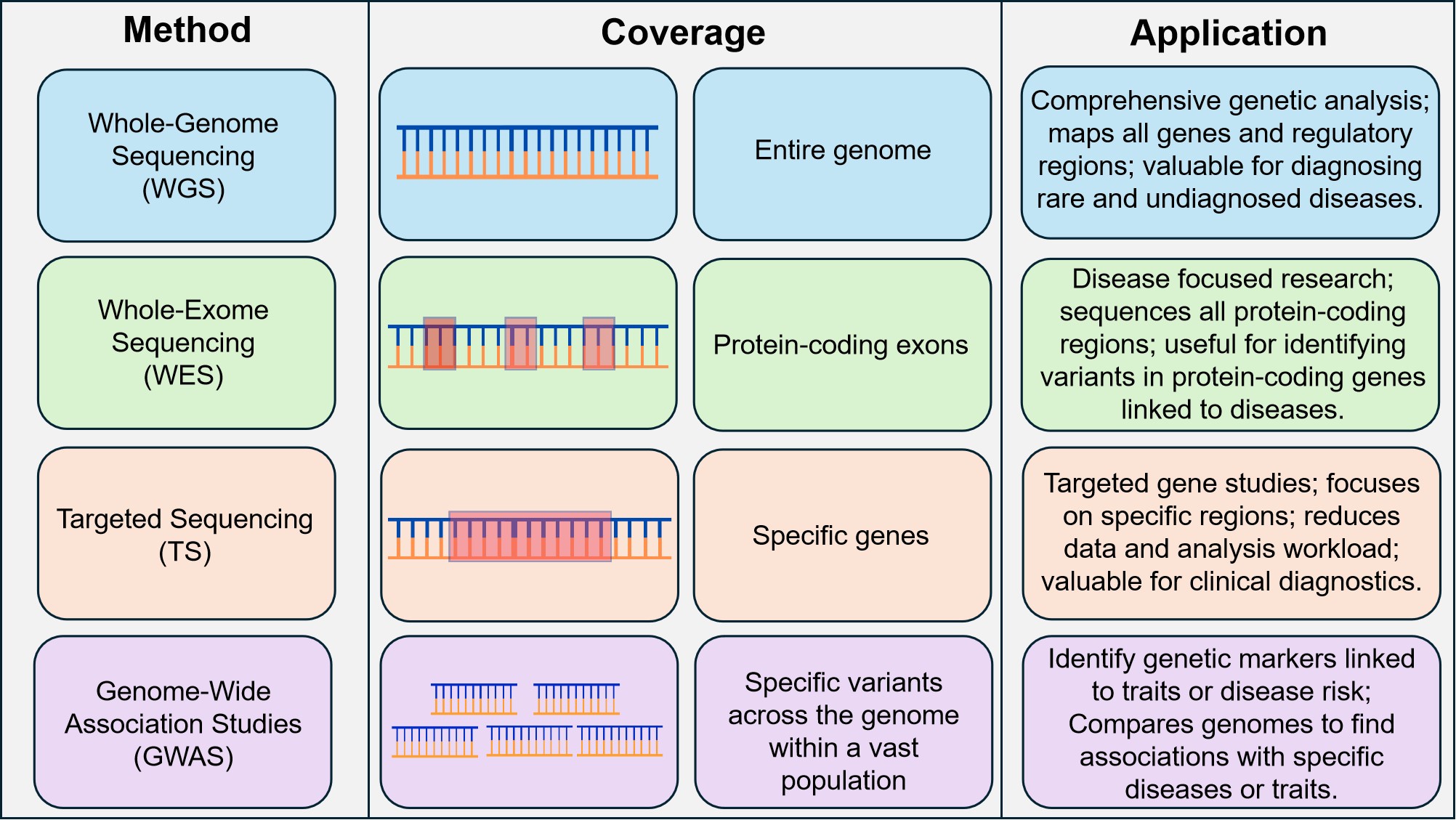
Figure 5. Comparison of Four Major Genomic Sequencing Techniques — WGS, WES, TS, and GWAS — Highlighting Their Applications and Benefits. WGS provides comprehensive coverage of the genome, WES focuses on protein-coding regions, TS targets specific genes for efficiency, and GWAS identifies genetic markers across populations. These methods are pivotal in diagnosing and understanding both common and rare neurological disorders.
Depending on the platform used, sequence output, referred to as reads, can range in size from ~500bp to >2Mb[57]. While short-read sequencing is relatively cost-effective and accurate[58], long-read sequencers offer significant benefits such as facilitating de novo assembly, detecting structural variants, and reducing amplification-induced bias[59,60]. In general, long-read sequencing is preferable for genome assemblies[57], whereas short-read sequencing is better for mutation detection. Herein, we highlight some key advances in our understanding of neurological disorders, mediated by next generation sequencing.
3.1 WGS
WGS is a comprehensive method for analysing an individualʼs entire genetic blueprint by sequencing all of the DNA in a sample[61]. WGS maps all genes and regulatory regions across the genome, uncovering variances including insertions, deletions, and single nucleotide polymorphisms (SNPs). For example, a recent study on PD revealed that small genomic deletions are associated with a higher risk of developing the disease, while small genomic gains are linked to a lower risk[62].
WGS maps all genes and regulatory regions across the genome, uncovering variances including insertions, deletions, and SNPs that comprise their unique genetic signature, making WGS a particularly powerful tool for identifying genetic causes of diseases with complex genetic architectures[63]. This technique is particularly valuable in clinical settings for diagnosing rare and undiagnosed diseases whilst capturing most genomic variations without the need for sequential genetic testing[64]. Unlike TS and WES, which focus on specific genes or exons, WGS covers the entire genome, including all genes and non-coding regions[63]. The WGS workflow is streamlined and less labour-intensive compared to other methods, such as TS, due to the absence of the capture and amplification steps[61]. Additionally, robust computational infrastructure is needed to handle the large volume of data generated by WGS, and quality control is crucial throughout the process[61,64].
Recently, Oh et al.[62], described the use of WGS to identify small genomic deletions, gains, and SNVs, and their association with an increased risk of developing PD (outlined in Figure 6). PD, the second most prevalent neurological ailment, is characterised by motor dysfunctions such as tremors, rigidity, bradykinesia, and postural instability and is complicated and impacted by both environmental and hereditary variables[65,66].
|
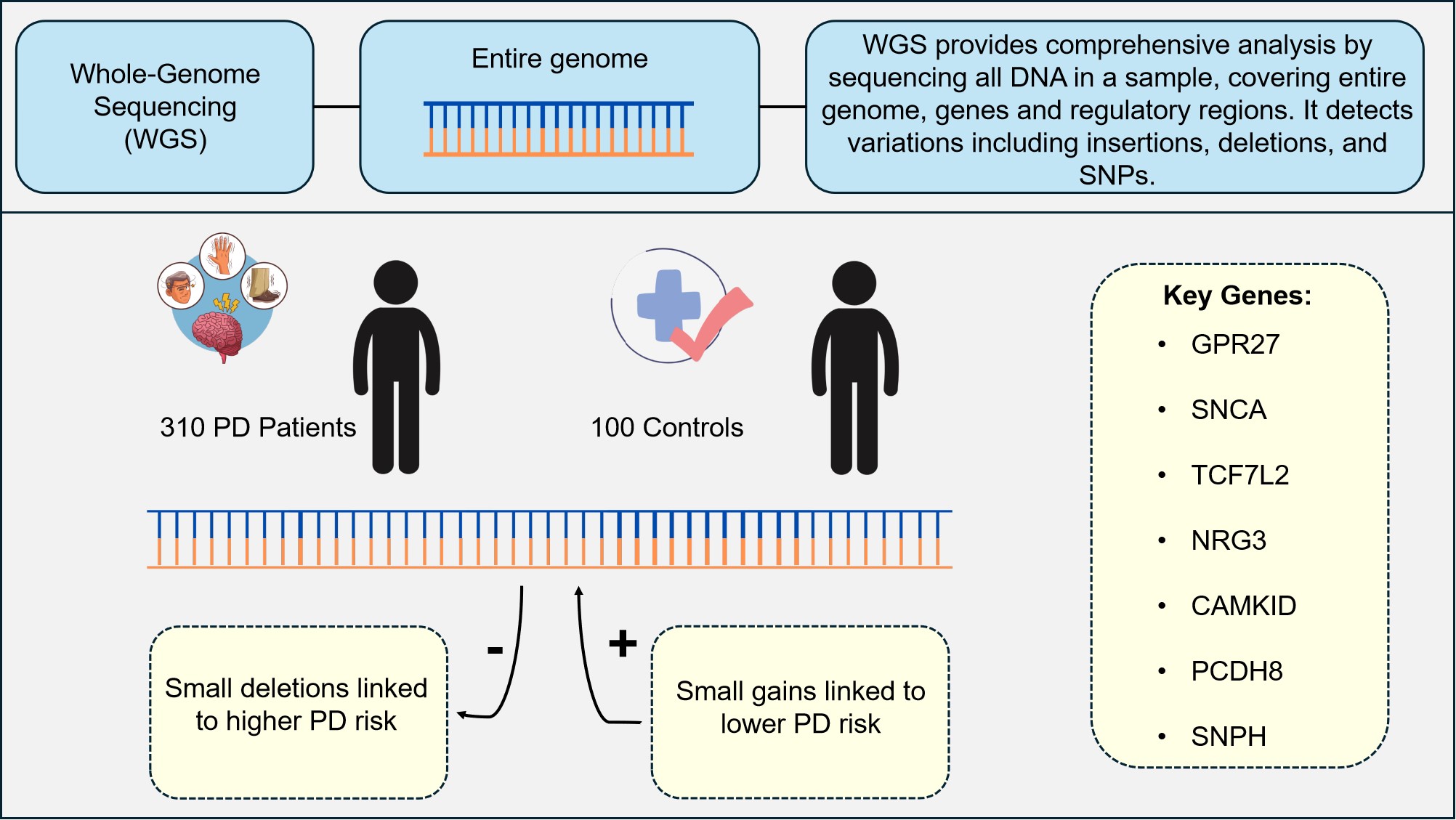
Figure 6. WGS Analysis of PD Patients and Controls. This study utilised WGS to analyse 310 patients with sporadic PD and 100 healthy controls. The analysis revealed that small genomic deletions are associated with a higher risk of PD, while small genomic gains are linked to a lower risk of developing the disease. Key genes identified in the study include GPR27, SNCA, TCF7L2, NRG3, CAMK1D, PCDH8, and SNPH. These genetic alterations influence PD risk through mechanisms such as downregulation of dopamine neurotransmitter release and upregulation of SNCA expression. The findings were further validated using an additional cohort of PD patients and controls, highlighting the importance of these genomic variations in PD susceptibility[62].
The study by Oh et al.[62], focused on 310 Korean patients with sporadic PD, and 100 healthy controls. WGS data from a separate secondary cohort (made up of 100 patients with sporadic PD and 100 healthy controls) was used to validate the results. High-read-depth WGS (average depth of 54×) allowed for a comprehensive analysis of germline variants, including SNVs, insertions/deletions (InDels), and copy number variations (CNVs). The results were robust, with the primary and secondary cohorts showing consistent findings.
Global minor genomic deletions were linked to a higher risk of developing PD, according to high-read-depth WGS data. Specifically, 30 significant locus deletions were identified, most of which were associated with an increased risk of PD in both cohorts. Global small genetic gains, on the other hand, were linked to a lower likelihood of PD onset (Figure 7).
|
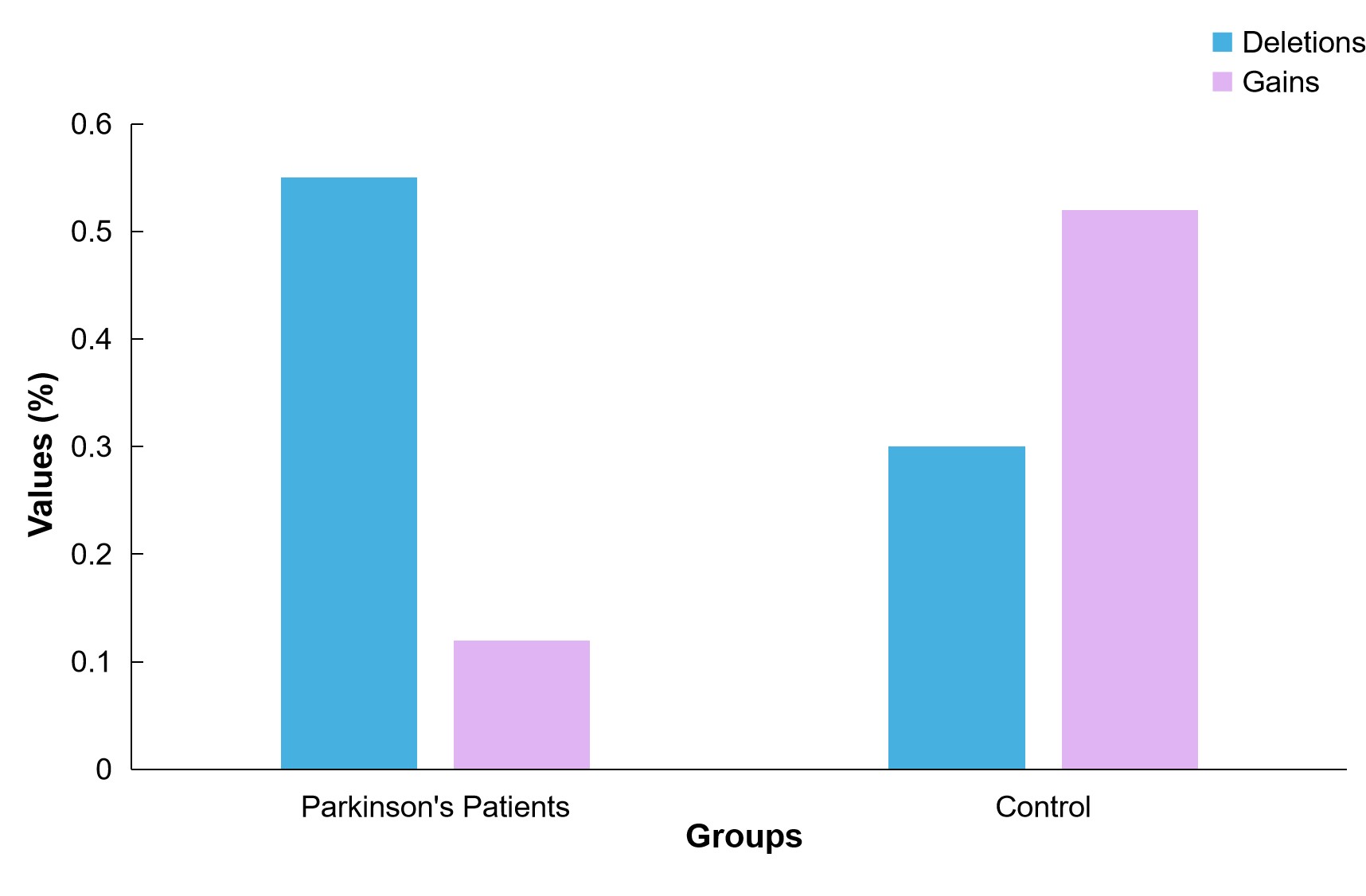
Figure 7. A Conceptual Representation of the Frequency of Small Genomic Deletions and Gains in PD Patients Versus Controls. Small genomic deletions were significantly more frequent in PD patients, while gains were more common in controls.
Additionally, it was observed that PD patients had clustered minor genomic deletions in the GPR27 region, resulting in a downregulated dopamine neurotransmitter release cycle, and elevated SNCA expression. A key factor in the validation of these results was statistical analysis. The Wilcoxon rank-sum test was used to assess differences between continuous variables, such as sequencing depth and ages among cohorts, whereas Fisherʼs exact test was employed in the study to detect significant CNV areas. Significant SNVs associated with PD, such as those in the NRG3 and CAMK1D genes, which were further supported by functional validation using data from the GTEx and Cancer Cell Line Encyclopedia (CCLE) databases. Additionally, a Sequence Kernel Association-Optimized (SKAT-O) analysis was performed to investigate rare missense and pathogenic mutations, identifying PCDH8 and SNPH as genes significantly associated with PD.
Strong evidence for the genetic contributions to PD is provided by the complete strategy that integrates WGS data with rigorous statistical analysis and functional validation. Results point to meaningful contributions from particular genomic deletions and SNVs, especially those impacting regulatory areas, toward the risk of developing PD.
3.2 WES
WES has become an invaluable clinical tool in the discovery of uncommon genetic variations linked to neurological illnesses[67]. Given that WES is particularly suited to short reads, making it cost-effective, clinicians are better able to investigate the composition, genetic polymorphisms, and roles of different genome variations within certain populations[71]. This approach thus holds significant promise for improved diagnosis, prevention, and treatment of neurological disorders[69,70].
A recent application of WES in neurological disorders was performed by Alvarez-Mora et al.[72], involved a retrospective study of 209 patients with a clinical diagnosis of neurodevelopmental, neurological, or neurodegenerative illnesses, including ataxia (40), spastic paraplegia (34), dystonia (46), Parkinson (23), intellectual disability (8), autism spectrum disorder (23), epilepsy (23), and other (15) (Figure 8A). An overall diagnostic yield of 32% was achieved, identifying disease-causing variants in 66 patients.
|
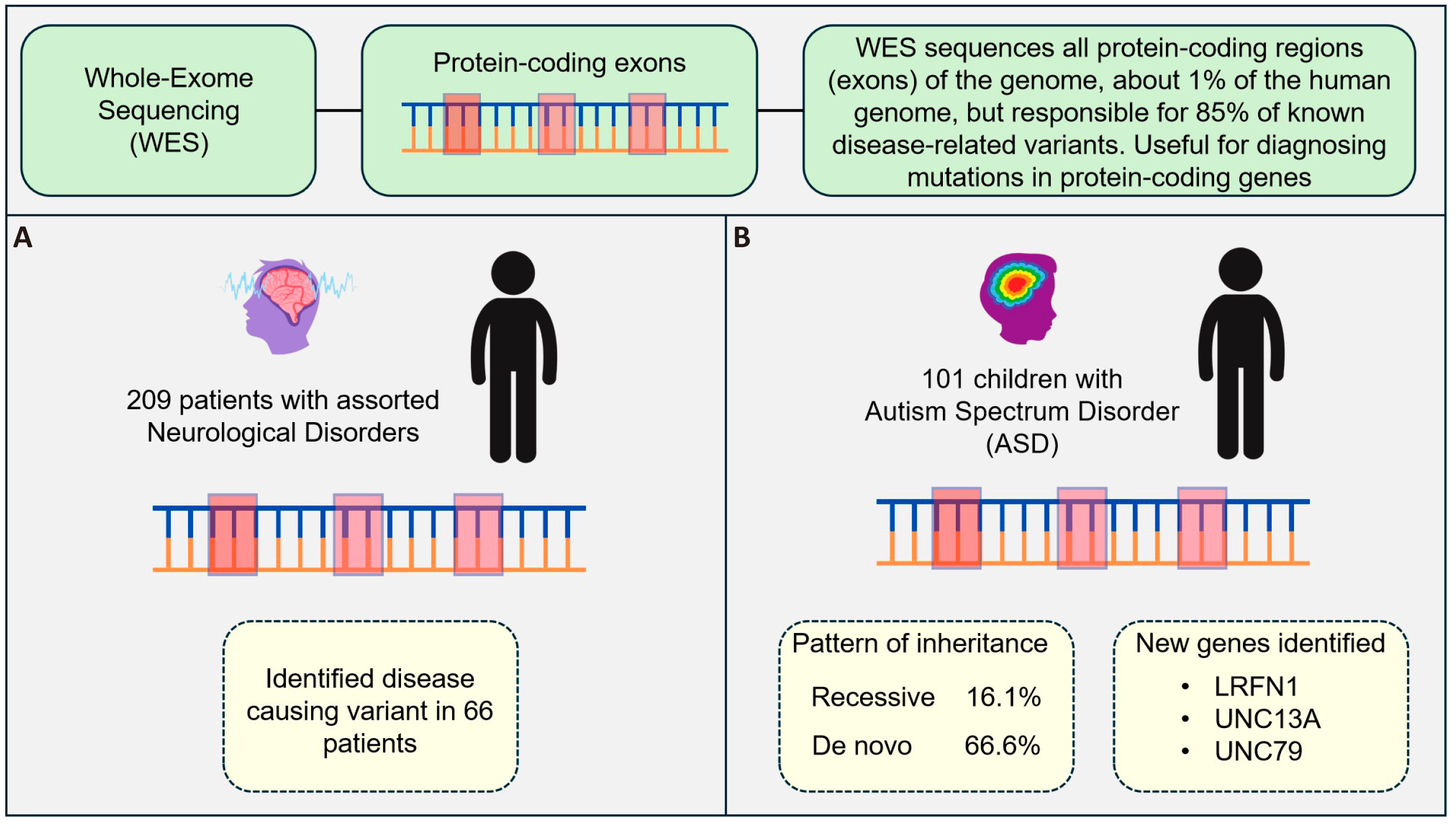
Figure 8. WES Overview. WES sequences the protein-coding regions of the genome, which constitute about 1% of the human genome but account for 85% of known disease-related variants. It is a valuable tool for diagnosing mutations in protein-coding genes. A) In a retrospective study by Alvarez-Mora et al.[72], involving 209 patients with assorted neurological disorders, disease-causing variants were identified in 66 patients, achieving a 32% diagnostic yield. B) In a study of 101 children with ASD by Sheth et al.[73], new genes were identified, and the pattern of inheritance of pathogenic genes was determined. The study found that 16.1% of the identified variants were inherited recessively, while 66.6% were de novo mutations. New genes linked to ASD, such as LRFN1, UNC13A, and UNC79, were also discovered, underscoring the importance of WES in understanding the genetic basis of ASD.
Significant differences in diagnostic yield were observed across disorders, with spastic paraplegia showing the highest yield at 64.7%, and dystonia the lowest at 15.2%. Chi-square tests demonstrated the statistical significance of these discrepancies and the variable efficacy of WES under various circumstances. Comparative analysis with other studies showed that the diagnostic yield for spastic paraplegia in this study (64.7%) was significantly higher than the 40% reported in other research, strengthening the credibility of the results.
Based on these results, the authors concluded that applying WES in clinical routine care would not only benefit patients, but also their families, based on its ability to estimate disease risk, discover aetiology and, in some cases, to identify specific treatment options.
In support of this, Sheth et al.[73], demonstrated the benefits of WES when compared to karyotyping, FMR1 triplet repeat expansion, and chromosomal microarray, for identifying the genetic architecture of autism spectrum disorder (ASD) (Figure 8B). ASD affects approximately 1 in 160 children worldwide, and is linked with difficulties in social communication, together with repetitive and obsessive behaviours, and/or limited interests that may persist over a lifetime. The aetiology of ASD remains unclear, though its similarity to other neurodevelopmental disorders suggests that genetics and environmental influences may contribute to its pathogenesis[74]. Based on an analysis of 101 Indian children with confirmed clinical diagnosis of ASD, the findings of Sheth et al.[73], strongly supports the use of WES as a key genetic diagnostic technique for ASD. Compared to 2.9%, 0%, and 0% from CMA, FMR1 triplet repeat expansion, and karyotype testing, respectively, WES identified pathogenic/likely pathogenic mutations responsible for the ASD phenotype in 29.7% of cases. All three of the CNVs found by CMA were also identified by WES, along with a fourth that was solely found by WES. Of the individuals with a genetic diagnosis of ASD, the pattern of inheritance for the variant was found to be recessive in 16.1% of cases and de novo in 66.6%. Three genes associated with ASD have been newly identified: LRFN1, UNC13A, and UNC79 with a spontaneous occurrence resulting in the alteration of the LRFN1 gene. The interaction between LRFN1 and DLG4, a recognized ASD gene, is responsible for forming the post-synaptic complex that facilitates signal transmission. Due to their close association, it is not unreasonable to consider LRFN1 to be a potential candidate for ASD; however, substantiating functional evidence is still required. The variants found in both UNC13A and UNC79 genes were categorized as variants of uncertain significance (VUS). These variations were inherited from parents who are likely asymptomatic carriers. As there was no correspondence between these genetic changes and any documented phenotypes within the OMIM database, they were included in both AutDB and SFARI databases as novel findings.
And Figure 9 summarises findings from the studies by Alvarez-Mora et al.[72] and Sheth et al.[73], comparing the effectiveness of WES in diagnosing spastic paraplegia, dystonia, and ASD, highlighting WESʼs effectiveness in diagnosing these conditions.
|
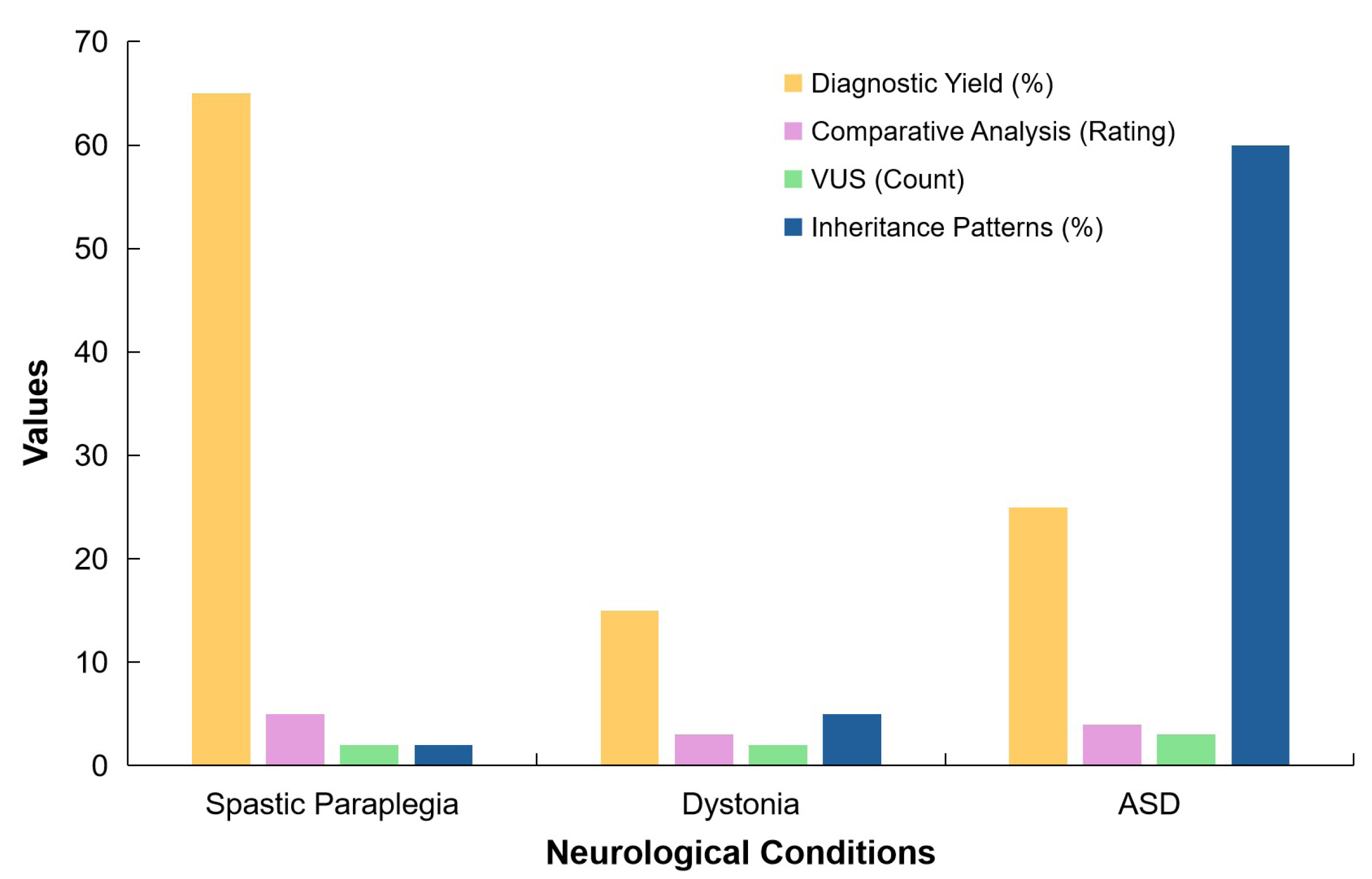
Figure 9. A Conceptual Representation of Key Metrics Across Three Neurological Conditions: Spastic Paraplegia, Dystonia, and ASD. The chart illustrates the percentage of cases with a diagnostic yield from clinical exome sequencing, with Spastic Paraplegia showing the highest yield. It also represents comparative analysis ratings, indicating the relative difficulty in analysing these conditions. The chart shows the count of VUS, reflecting the complexity of genetic interpretation in each condition. Finally, it depicts the percentage of inheritance patterns identified, with ASD having a significantly higher percentage. This chart highlights the variability in diagnostic outcomes and genetic findings across different neurological conditions.
3.3 TS
TS, sometimes referred to as target enrichment sequencing, is a highly specialised and cost-effective genomic sequencing technique that involves targeting specific regions of the genome, such as genes, exons, or other areas of interest, rather than an entire genome or exome[66,75,76]. This approach offers a more efficient alternative by focusing on relevant sections, thereby reducing the amount of data generated and the subsequent analysis workload[75]. TS is particularly valuable in clinical diagnostics for identifying mutations or variants that can quickly explain a patientʼs condition[75] (Figure 10). Additionally, it is widely used in both research and clinical settings to detect mutations in known disease-associated genes and to understand the genetic basis of diseases[76].
The two principal methods involved in TS are amplicon sequencing and hybridization capture. The process of amplicon sequencing involves the utilisation of specially designed primers that are employed during PCR to amplify specific regions within the genomic DNA. Hybridization capture on the other hand commences with an NGS library and establishes connections between probes and molecules in the library possessing the necessary sequence. Following the separation of the resulting complexes, enriched samples are generated for subsequent amplification and sequencing. Compared to amplicon sequencing, although more intricate, hybridization capture offers the advantage of enhanced sensitivity, improved uniformity, reduced occurrence of PCR artefacts, and enables simultaneous assessment of millions of targets[52,55].
|
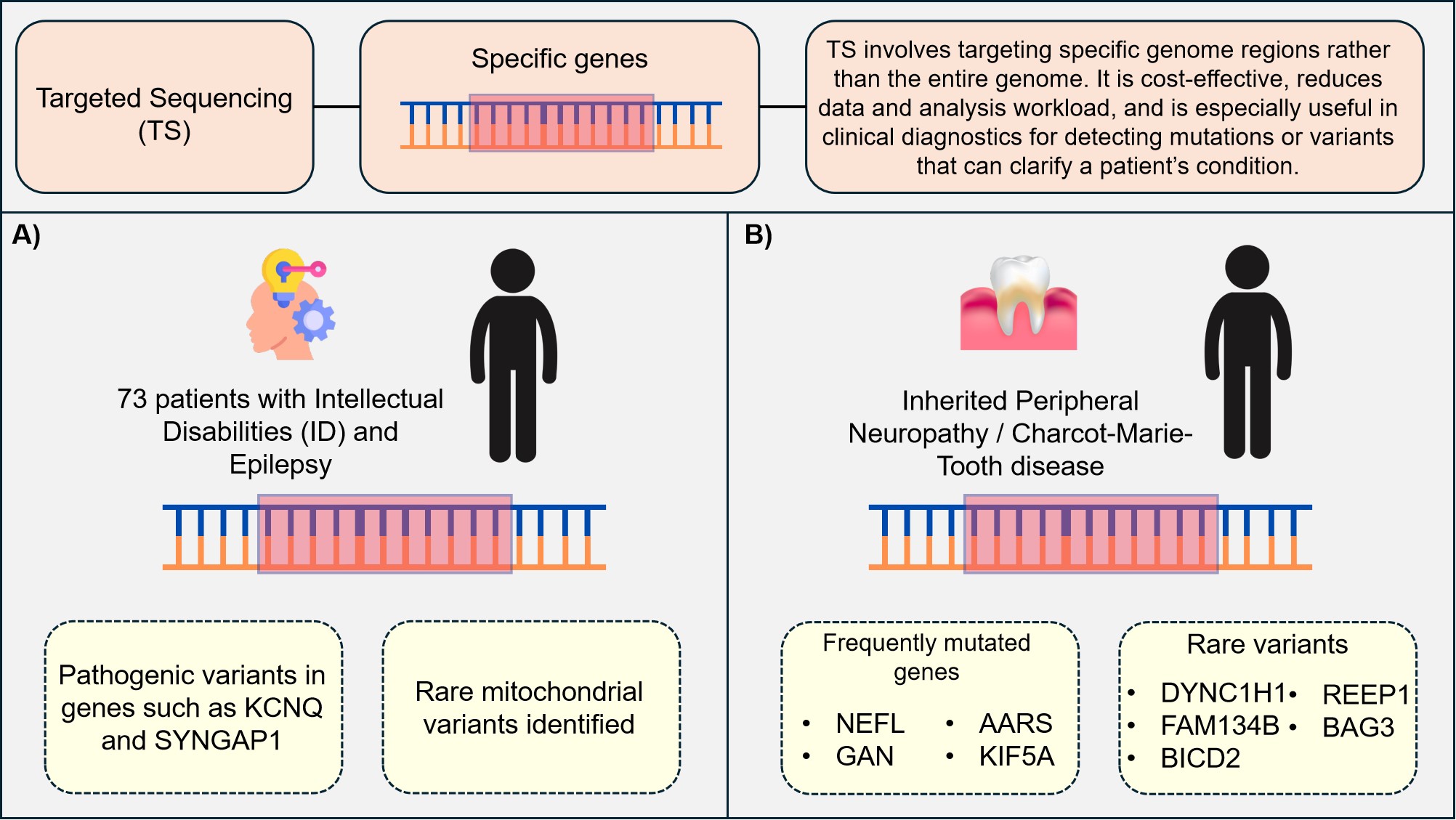
Figure 10. TS in Clinical Diagnostics.
TS focuses on specific regions of the genome, making it a cost-effective and efficient method for clinical diagnostics. It is particularly useful for detecting mutations or variants that can clarify a patient’s condition. As illustrated in Figure 10. A) In a study of 73 patients with intellectual disabilities and epilepsy, TS identified pathogenic variants in genes such as KCNQ and SYNGAP1, along with rare mitochondrial variants[50]. B) In the case of inherited peripheral neuropathy (IPN) and CMT, TS revealed frequently mutated genes such as NEFL, GAN, AARS, and KIF5A, as well as rare variants like BAG3, BICD2, DYNC1H1, REEP1, and FAM134B[77]. These findings demonstrate the effectiveness of TS in identifying both common and rare genetic mutations associated with these conditions, providing valuable insights for diagnosis and treatment.
TS has been used successfully to analyse individuals with intellectual disabilities (ID)[50]. Intellectual functioning and adaptive behaviour are limited in individuals with ID[78]. Beginning at birth, these limitations fully manifest by the age of 18 and can be associated with a wide range of co-occurring conditions. These conditions can include neurological disorders (e.g., epilepsy), mental health conditions (e.g., depression and anxiety), and other medical conditions (e.g., meningitis)[79,80]. The most recent gene panels for ID used in diagnostic laboratories contain around 1,500 genes[81]. Skoczylas et al.[50] used a panel of targeted NGS to search for pathogenic variations in genes linked to the onset of moderate to severe ID and/or epilepsy in the nucleus DNA (nuDNA) and pathogenic mitochondrial DNA (mtDNA). The approach involved a cohort of 73 patients, comprising 34 individuals with ID or developmental delay, 21 with epilepsy, and 19 with a combination of both conditions. Genetic material was obtained from peripheral blood, and a specialized gene panel was employed for library preparation using the Agilent SureSelectQXT Target Enrichment protocol. Subsequently, paired-end sequencing was conducted on an Illumina NextSeq550 System. Several pathogenic and likely pathogenic variants within different genes linked to epilepsy and intellectual disability were found over the course of the investigation. Most notably, de novo mutations were observed in genes such as KCNQ and SYNGAP1 which contribute to the development of autosomal dominant disorders. Furthermore, the analysis uncovered a set of rare heteroplasmic mitochondrial variants, some of which are linked to established pathogenic variants, including m.5,521G>A and m.7,947A>G. However, within the overall cohort of 73 patients, only nine demonstrated distinctly causative variants, thus illustrating the inherent complexity in the diagnosis of these disorders.
With a prevalence of one in 2,500, CMT is a major cause of neurological disability, marked by extensive genetic heterogeneity[82]. Prior to the development of NGS, CMT screening relied on Sanger sequencing of candidate genes. However, the advent of TS, specifically a panel of 81 IPN/CMT genes, has significantly improved the molecular diagnosis of this condition. A seminal study of IPN, by Bacquet et al.[77], focused on the three main categories of CMT, hereditary sensory and autonomic neuropathy (HSAN), and distal hereditary motor neuropathy (dHMN).
In this study, 123 unrelated patients with diverse forms of IPN underwent targeted NGS. The molecular diagnosis was resolved in 49 of the 123 patients (~40%). Panel-based NGS was particularly effective in familial cases, with a diagnostic yield of 49%, compared to 19% in sporadic cases. Additionally, NGS-based screening identified three CNVs, further raising the diagnostic yield to 41%, which is two times higher than the previously used Sanger sequencing strategy.
Statistically, the study used a two-tailed Fisher’s exact test to compare the diagnostic yield between the targeted NGS strategy and the previous Sanger sequencing approach in a retrospective cohort of 56 patients. This analysis confirmed the significant improvement provided by the NGS approach. Among the identified variants, 26 were previously reported in the literature, while 52 were novel. Pathogenic variants were confirmed in 49 patients (40%), and potentially pathogenic variants were identified in an additional 11 patients (9%).
Notably, several genes (MFN2, SH3TC2, NEFL, GAN, DGAP1, AARS, and KIF5A) were found to be frequently mutated, with NEFL, GAN, AARS, and KIF5A having a higher frequency of pathogenic variants. Even rare variants were found in BAG3, BICD2, DYNC1H1, REEP1, and FAM134B, proving the value of NGS in improving molecular diagnostics and finding mutations that had not been found before.
While Figure 11 provides a comparative analysis of diagnostic yield, notable genetic findings, and the effectiveness of TS across two studies focused on intellectual disabilities/epilepsy and CMT.
|
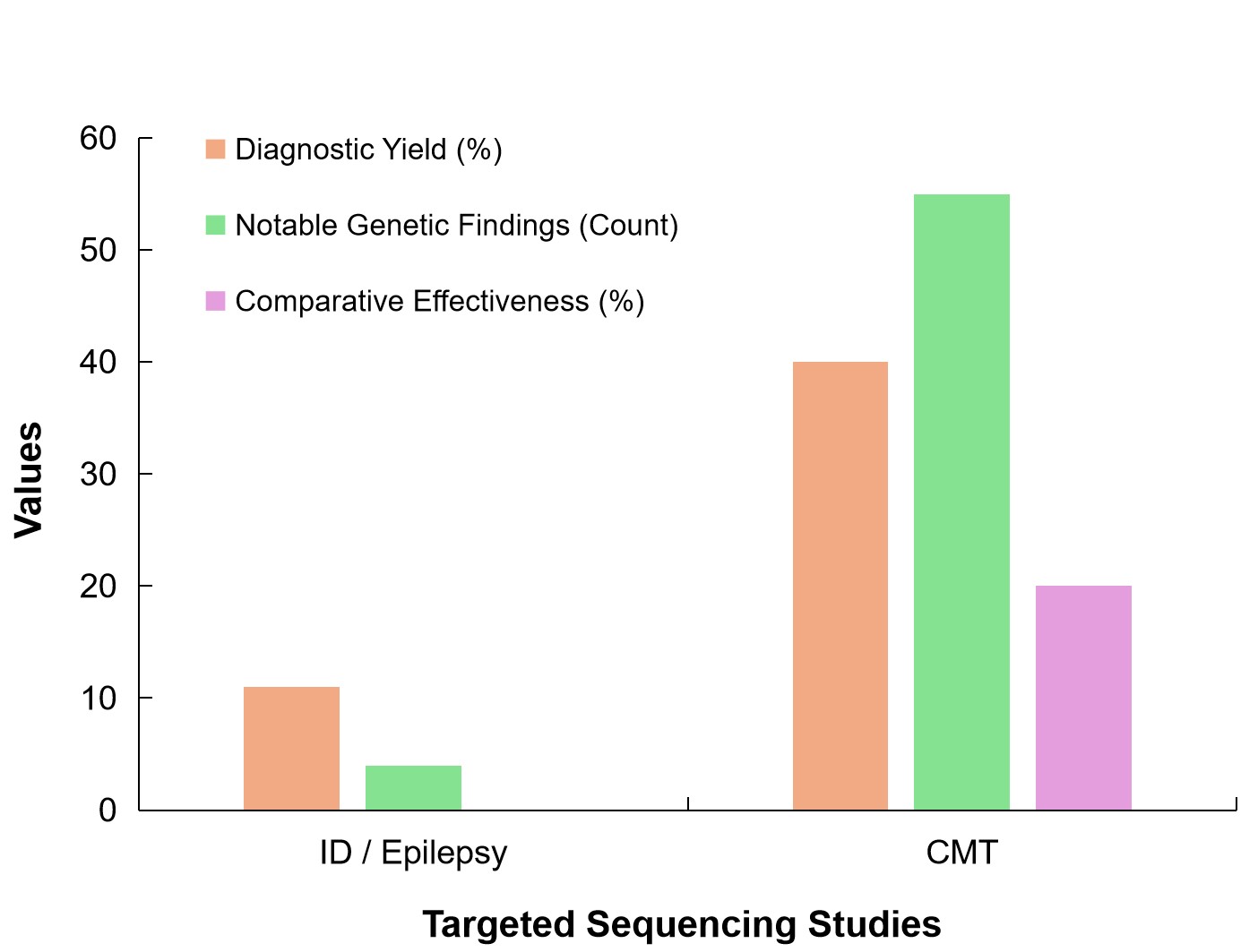
Figure 11. The Bar Chart Compares Two Studies on TS in Terms of Diagnostic Yield, Notable Genetic Findings, and Comparative Effectiveness. The ID/Epilepsy study (Skoczylas et al.[50]) shows a diagnostic yield of 12%, with 4 notable genetic findings, including de novo mutations and mitochondrial variants. The CMT study (Bacquet et al.[77]) demonstrates a higher diagnostic yield of 40%, with 55 notable genetic findings, including CNVs and novel variants. The chart also highlights the 20% improvement in diagnostic yield achieved through TS compared to other methods in the CMT study.
3.4 GWAS
GWAS are designed to identify genetic variants linked to specific diseases or traits by comparing allele frequencies between phenotypically different groups[35,83,84]. The method involves scanning the entire genome of numerous individuals to find SNPs that are more frequent in individuals with a particular disease compared to those without[84,85]. In GWAS, common genetic variations associated with certain illnesses are found by examining the genomes of large cohorts[86]. By analysing the relationships between several genes, GWAS has significantly improved our understanding of the genetic basis of many illnesses. This is especially beneficial for neurological disorders due to their complex nature and their interactions between various genetic factors[89,90].
GWAS test each genotyped or imputed variant across the whole genome sequentially within a regression framework, identifying genetic variants that meet the stringent threshold for genome-wide significance (P<5e-8) as being strongly linked to the trait or disease being studied[83]. This approach allows researchers to study the entire genome without needing a predefined hypothesis about the gene locations involved[84]. Additionally, GWAS often identifies associations between traits or diseases and groups of genetic variants that are inherited together due to their physical proximity on the chromosome, a phenomenon known as linkage disequilibrium (LD), which in turn motivates further detailed analyses to prioritise and uncover the specific genes that are causally implicated in the phenotype[83].
Several important genetic contributors including APOE, GBA, GRN, LRRK2, MAPT C9orf72, and others, have previously been linked to pleiotropic effects across different neurodegenerative diseases (NDDs). These diseases affect millions globally and include such conditions as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). In a recent study by Koretsky et al.[91], GWAS was used to examine common genetic variations linked to an increased risk of NDD across a sample size of 5,000, chosen from amongst 23,885, grouping individuals according to their genetic profiles and risk factors (Figure 9A). To enable comprehensive multi-disease and disease-specific analysis, this cohort includes instances of frontotemporal dementia, PD, AD, amyotrophic lateral sclerosis, and Lewy body dementia.
To group individuals according to their genetic risk profiles, the study used GWAS summary statistics and genome-wide SNPs. After rigorous quality control, including ancestry verification and pruning, 338 GWAS-significant SNPs were analyzed using Unified Manifold Approximation and Projection (UMAP) for dimensionality reduction, followed by unsupervised clustering via the mean shift algorithm. This approach identified three primary clusters, with Cluster 0 enriched for ALS cases (OR=1.631, P=4.66×10-8), Cluster 1 for AD (OR=1.637, P=9.20×10-9), and Cluster 2 for frontotemporal dementia (OR=3.063, P=6.50×10-5). Figure 12 presents the clustering of genetic risk factors for various neurodegenerative diseases, identifying distinct groupings of individuals based on their genetic predispositions to ALS, AD, Lewy body dementia, and frontotemporal dementia.
|
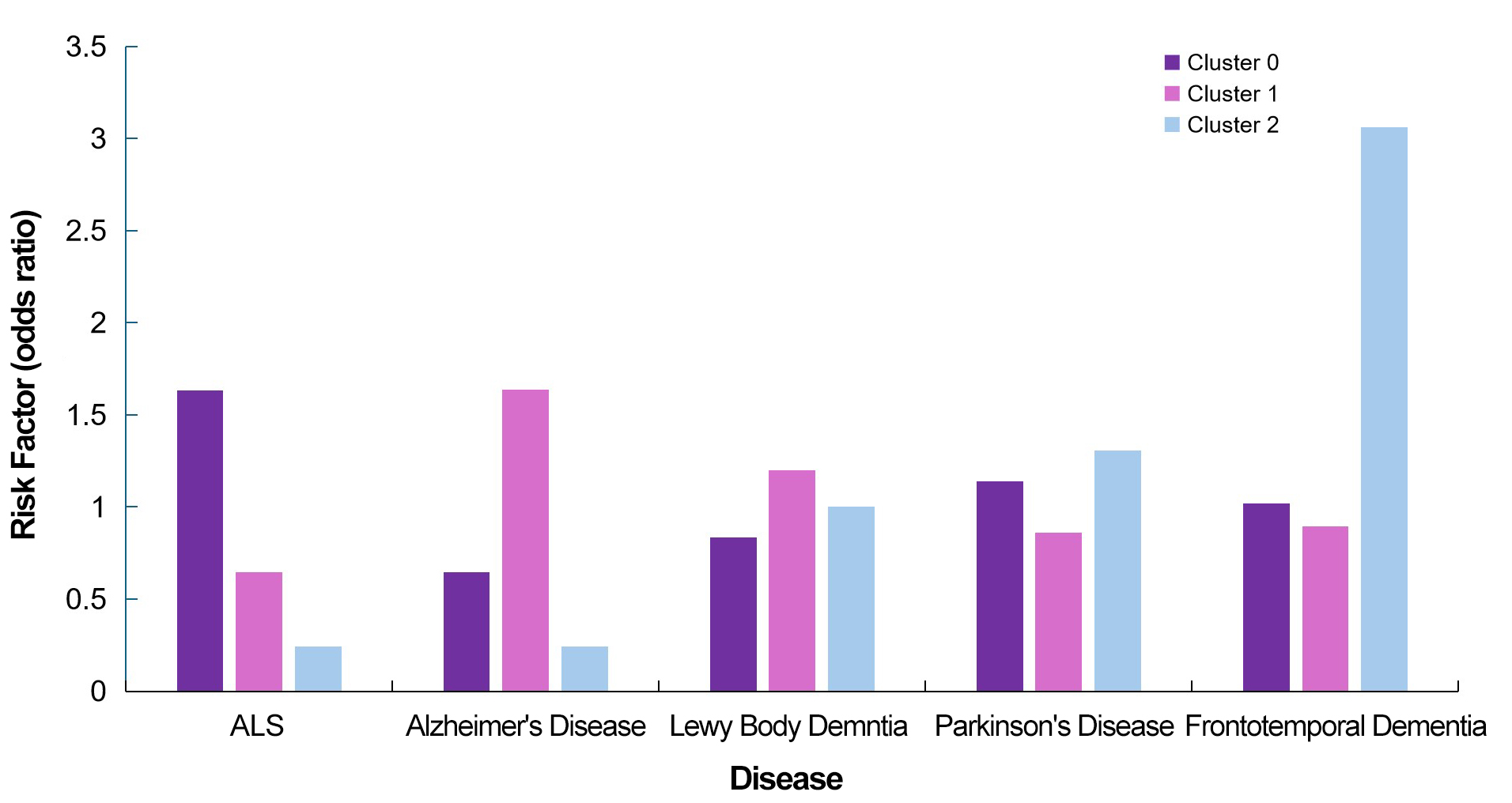
Figure 12. Illustrates the Genetic Risk Factors (Odds Ratios) Associated with Various Neurodegenerative Diseases (ALS, AD, Lewy Body Dementia, PD, and Frontotemporal Dementia) Across Three Identified Genetic Clusters (Groups of Individuals Classified Based on Their Genetic Profiles). Each disease is represented by three bars, corresponding to Cluster 0, Cluster 1, and Cluster 2. The height of each bar indicates the Odds Ratio (OR), reflecting the strength of association between that cluster and the disease. Higher OR values suggest a stronger genetic predisposition within that cluster. For example, Frontotemporal Dementia shows the highest risk in Cluster 2, whilst ALS is most strongly associated with Cluster 0. This visualisation helps to compare the distribution of genetic risks across different diseases and clusters.
An important finding was that individuals diagnosed with one NDD often had higher genetic risk for others. For instance, the polygenic risk score (PRS) for Lewy body dementia was strongly associated with genetic risk across multiple NDDs. The significance of discovering these loci for precise diagnosis and clinical trial design is highlighted by the overlap in genetic risk loci across several neurodegenerative diseases (Figure 13).
The study’s results were validated through multiple comparative analyses, confirming the robustness of the clustering approach and its implications for understanding the genetic interconnectivity of NDDs.
|
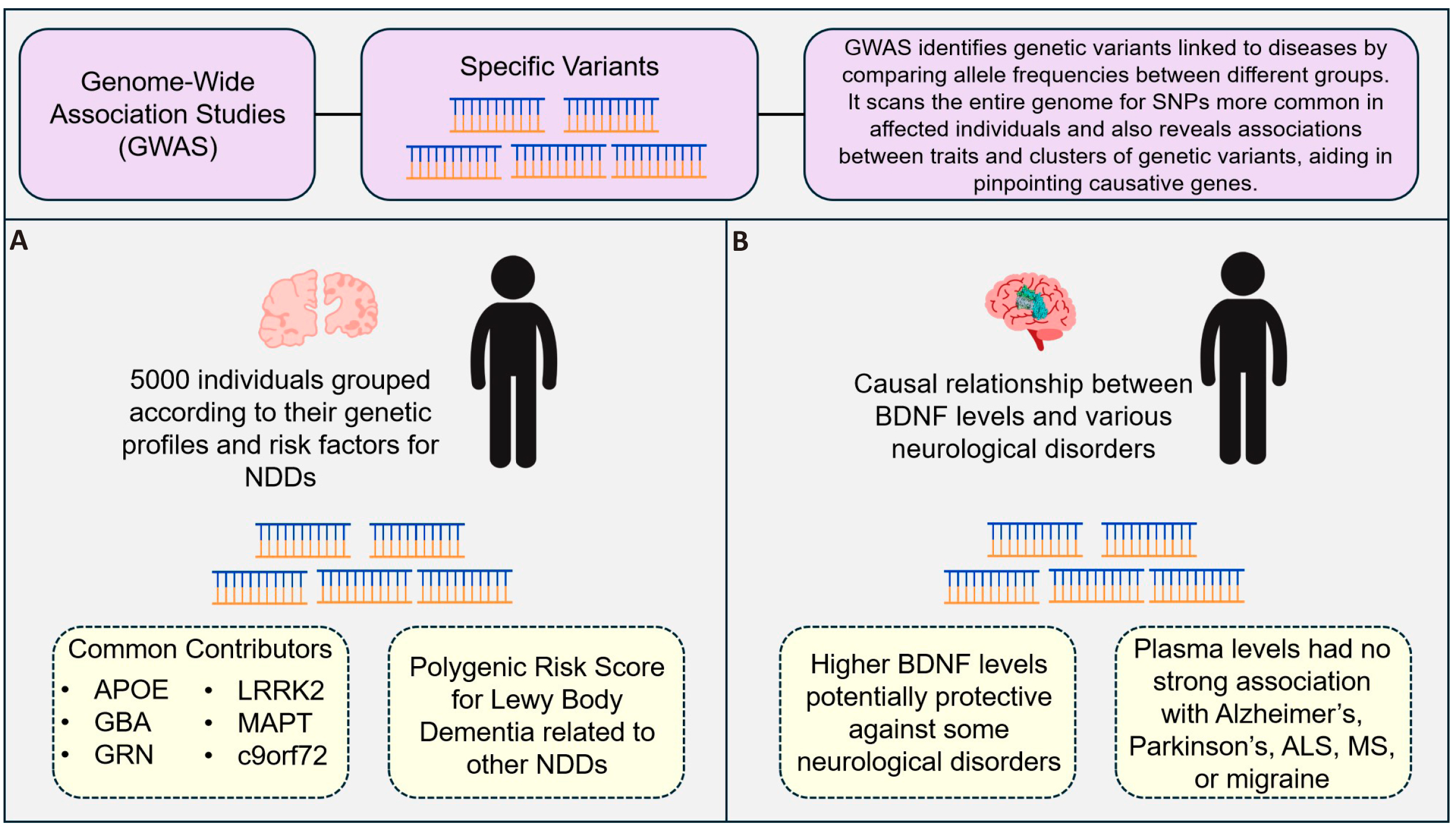
Figure 13. GWAS in Identifying Genetic Variants Linked to Neurodegenerative Diseases (NDDs). GWAS identify genetic variants associated with diseases by comparing allele frequencies between different groups, scanning the genome for SNPs more common in affected individuals. This process also reveals associations between traits and clusters of genetic variants, helping to pinpoint causative genes. A) A study of 5,000 individuals grouped according to their genetic profiles and risk factors for NDDs revealed common genetic contributors, such as APOE, GBA, GRN, LRRK2, MAPT, and C9orf72. Additionally, a polygenic risk score for Lewy body dementia was found to be related to other NDDs (Koretsky et al.[91]). B) An investigation into the causal relationship between Brain-Derived Neurotrophic Factor (BDNF) levels and various neurological disorders indicated that higher BDNF levels may be protective against some neurological conditions. However, no strong associations were found between plasma BDNF levels and conditions such as Alzheimerʼs, Parkinsonʼs, ALS, MS, or migraine[92].
The study by Wang et al.[92] (outlined in Figure 13B) represents a comprehensive research effort to uncover the potential causal relationships between plasma brain-derived neurotrophic factor (BDNF) levels and various neurological diseases using a Mendelian randomisation (MR) approach. This approach leveraged genetic variants as instrumental variables to infer causality, providing more robust evidence than traditional observational studies.
The researchers utilized GWAS data to select SNPs strongly associated with plasma BDNF levels. These SNPs served as instrumental variables in the MR analysis. Data on plasma BDNF levels were sourced from a large-scale GWAS including 3,301 individuals of European descent. For neurological disorders, the study included GWAS summary data from several large consortia: the MEGASTROKE consortium for stroke (40,585 cases and 406,111 controls), the FinnGen consortium for nontraumatic intracranial hemorrhage (nITH) (6,530 cases and 342,673 controls), and various consortia for neurodegenerative diseases such as AD, PD, amyotrophic lateral sclerosis (ALS), MS, as well as epilepsy and migraine.
The results of the MR analysis revealed that higher plasma BDNF levels are potentially protective against several neurological disorders. Specifically, higher plasma BDNF levels were associated with a reduced risk of nontraumatic intracranial haemorrhage (nITH). These findings were further supported by a combined analysis of data from three consortium datasets, which showed a significant protective effect of plasma BDNF on epilepsy and a suggestive impact on focal epilepsy.
Interestingly, no strong associations were found between plasma BDNF levels and other neurological disorders such as AD, PD, ALS, MS, or migraine. This indicates that the protective role of BDNF might be specific to certain neurological conditions, particularly those involving epilepsy and intracranial haemorrhage. Overall, this study underscores the potential of BDNF as a therapeutic target for specific neurological disorders. By integrating genomics with clinical and biochemical data through a multi-omics approach, the researchers provided new insights into the molecular mechanisms underlying these conditions. The use of Mendelian randomisation added a layer of rigor to the findings, highlighting the importance of genetic data in establishing causal relationships in medical research.
3.5 Advantages and Disadvantages of Each Genomic Technique for Sequencing
Each of the previously discussed methods (WGS, WES, TS, GWAS) has both strengths and weaknesses, making them suitable for different types of genetic studies and clinical applications. Herein, we provide a brief overview of these, with Table 4 providing a concise comparison of their main attributes and applications. Additionally, Figure 14 visually summarises the key metrics-such as comprehensiveness, diagnostic yield, cost-effectiveness, and complexity highlighting how WGS, WES, TS, and GWAS compare across these dimensions.
Table 4. Outlines the Advantages, Disadvantages and Potential Application of Various Genomic Sequencing Methods, Including WGS, WES, TS, and GWAS
|
Advantages |
Disadvantages |
Applications |
WGS |
Comprehensive data collection. High diagnostic yield. Detection of non-coding variants. Cost and time decrease. Longevity and reanalysis. Standardised workflows. |
Data volume and complexity. Risk of incidental findings. Data storage and privacy. Interpretation challenges. Ethical and social concerns. High cost. Data privacy. |
WGS is particularly valuable in understanding the genetic underpinnings of complex disorders like Parkinsonʼs disease by identifying small but significant genomic changes that contribute to disease susceptibility |
WES |
Cost-Effective. Higher Coverage. Effective in Identifying Variants. Diagnostic Yield. Useful in Complex Cases. Reduced Data Volume and Storage Needs. Powerful for Clinical Diagnostics. Ease of analysis. Better at Finding Rare Variants. |
Limited Scope. Misses structural and non-coding variants. Interpretation challenges. More expensive and complex than TS. Less comprehensive than WGS. Data reanalysis. |
WES is extensively used in clinical settings for neurodevelopmental and neurodegenerative disorders, allowing for a detailed analysis of protein-coding genes responsible for various neurological conditions. |
TS |
Cost effective. Reduced data generation. Reduced complexity. Clinically relevant. Specialised insights. Focused. |
Limited scope. Discovery limitation. Dependant on prior knowledge. |
TS focuses on specific genes known to be involved in intellectual disabilities, epilepsy, and inherited peripheral neuropathies, making it a practical tool in clinical diagnostics. |
GWAS |
Identification of genetic associations. Replication across populations. Large sample sizes. Diverse population analysis. |
Complexity and cost. Population representation bias. Focus on common variants. Need for further validation. Complexity of fine-mapping. Methodological challenges. |
GWAS are instrumental in identifying genetic markers associated with neurodegenerative diseases and are crucial for developing genetic risk profiles for complex conditions. |
Notes: This provides a comparative analysis to help researchers and clinicians choose the appropriate method based on their specific needs, considering factors like cost, scope, and diagnostic yield.
|
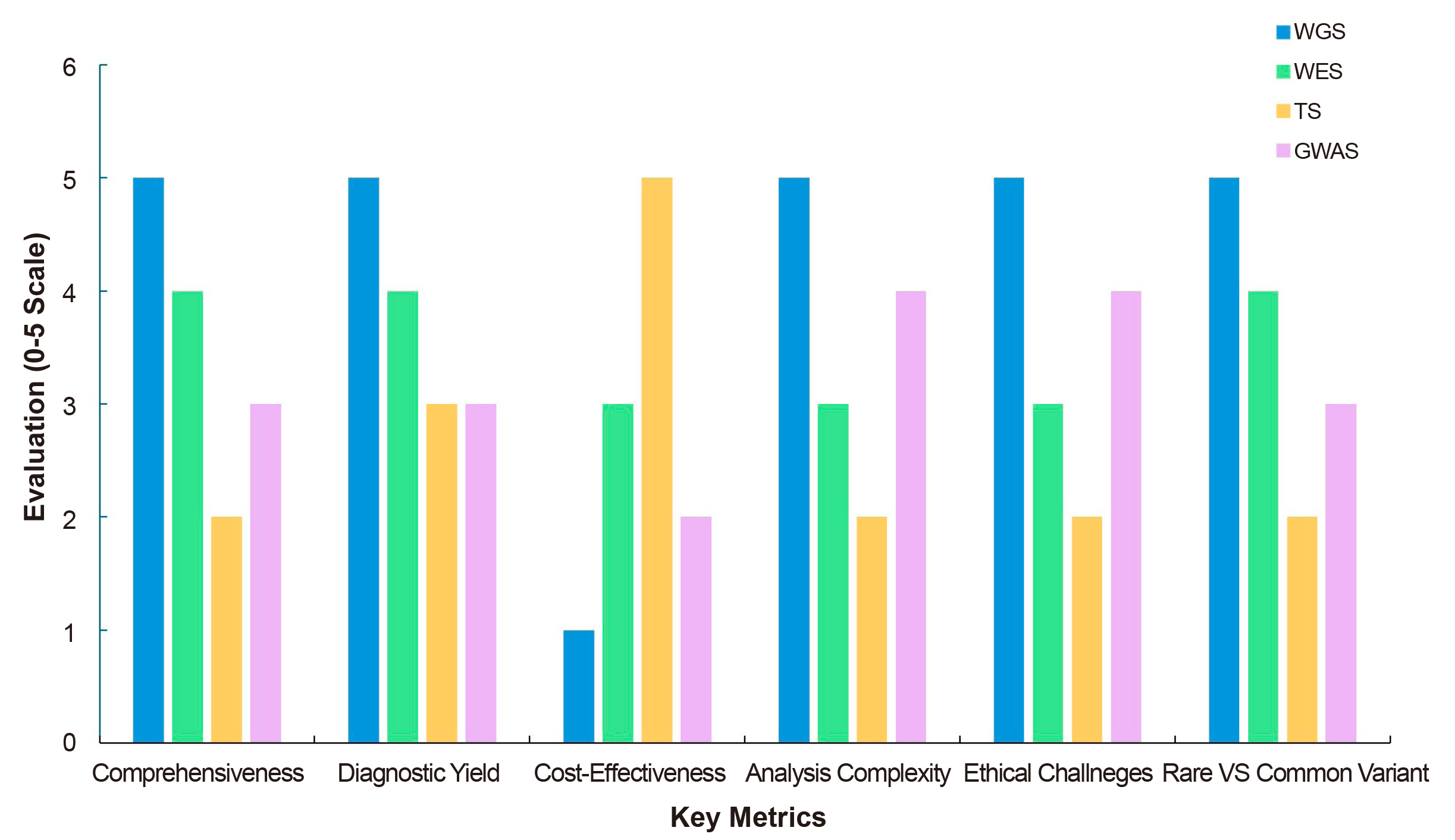
Figure 14. Conceptually Compares WGS, WES, TS, and GWAS Based on Six Key Metrics: Comprehensiveness, Diagnostic Yield, Cost-Effectiveness, Analysis Complexity, Ethical Challenges, and Usefulness for Rare vs. Common Variants. The evaluation is on a scale from 0 to 5, where higher scores indicate better performance or greater complexity.
WGS is a powerful tool in genetic diagnostics, offering several advantages, while also posing some challenges. WGS captures most genomic variations, including those in non-coding regions, providing a comprehensive data collection that surpasses panel or exome sequencing methods[63,64]. It has a high diagnostic yield, particularly in diagnosing rare and unknown diseases, as it can detect small somatic variants, CNVs, structural rearrangements, protein-coding variants, structural variations, non-coding variants, DNA repetition disorders, and mitochondrial mutations[63,64].
WGS also allows for reanalysis of data, making it a lifelong resource for patients as new clinical insights emerge[63,64]. Standardised workflows in WGS can minimize errors through accredited and automated protocols[64]. However, the vast amount of data generated by WGS necessitates a robust computational infrastructure, as well as specially trained staff for data processing and interpretation, which can be both costly and challenging[64]. Furthermore, the risk of incidental findings raises significant ethical concerns about patient consent and handling unexpected results[64]. Additionally, data storage and privacy are significant issues due to the large data files produced by WGS, requiring secure, often encrypted, data management practices to safeguard patient privacy[64]. The interpretation of variants also remains complex, with unresolved issues in variant classification and analytical challenges potentially leading to missed diagnoses[63,64].
WES, on the other hand, offers a more balanced approach between cost, coverage, and diagnostic yield compared to WGS, TS, and GWAS. WES is cost-effective because it focuses on exonic regions, resulting in less data generation and consequently easier analysis[67,69,70]. It also provides higher coverage of these regions, improving variant detection[67]. This method is particularly useful for identifying SNVs and small indels within protein-coding regions, essential for diagnosing complex phenotypes and rare Mendelian disorders[68]. However, WES does have some limitations, including missing non-coding regions, structural variants, and requiring specialized knowledge for variant interpretation[67,68]. When compared to WGS, WES is less comprehensive but more cost-effective and easier to manage due to smaller data volumes[68,70]. Although GWAS can identify common variants across the genome, WES is better at finding rare variants[67].
TS offers several advantages, including cost-effectiveness, efficiency, clinical relevance, and specialised insights. It is more cost-effective than WGS and WES as it focuses on specific genomic regions of interest, thereby reducing overall costs[55,75,76]. By generating less data, TS simplifies the analysis process and shortens the time required for data interpretation[75]. For this reason alone, TS is particularly beneficial in clinical diagnostics, providing precise information relevant to known disease-associated genes[75,76]. TS allows for a deep analysis of selected regions, leading to detailed insights into specific genetic variants and their implications[55,75]. However, it also has limitations, including a limited scope that may miss important variants outside of the targeted areas. This is particularly significant if unknown regions are relevant to the condition under investigation[75,76]. Additionally, it is less useful for discovering new genetic variations and for comprehensive genetic analysis since it does not provide information about the entire genome[75]. The success of TS relies heavily on prior knowledge of the genomic regions associated with specific diseases, making it less useful for conditions with a poorly understood underlying genetic bases[75]. Overall, TS is a focused, cost-effective approach for specific diagnostic and research applications, but lacks the comprehensive scope of WGS, WES, GWAS.
Finally, GWAS have significantly advanced our understanding of the genetic basis of various diseases by identifying numerous genetic variants associated with conditions like autism spectrum disorder (ASD), PD, and AD[83-85]. GWAS have been particularly effective in pinpointing SNPs and candidate genes, such as RELN, MECP2, and OXTR for ASD, across large sample sizes, increasing the credibility and statistical power of these findings[85]. Additionally, replication of GWAS findings across diverse populations further validates the associations and enhances the generalisability of the results[84,85]. The inclusion of diverse populations also helps uncover genetic variations specific to different ancestries, providing insights into global genetic architecture and facilitating fine-mapping efforts[84].
Despite these advantages, GWAS face several challenges. Conducting these studies requires large cohorts and extensive computational resources, making them resource-intensive[85]. The overrepresentation of certain populations can limit the generalisability of the findings to other groups and underscores the need for more inclusive studies[84]. Moreover, while GWAS are adept at identifying common genetic variants, they often miss rare variants and structural variations that contribute to disease risk, leading to the issue of missing heritability[83]. Further validation and replication in different cohorts are necessary to confirm the significance of GWAS findings[85]. Additionally, developing PRS from GWAS data, although useful for quantifying individual genetic risk, often suffers from poor transferability across different populations, limiting their broader application[83].
|
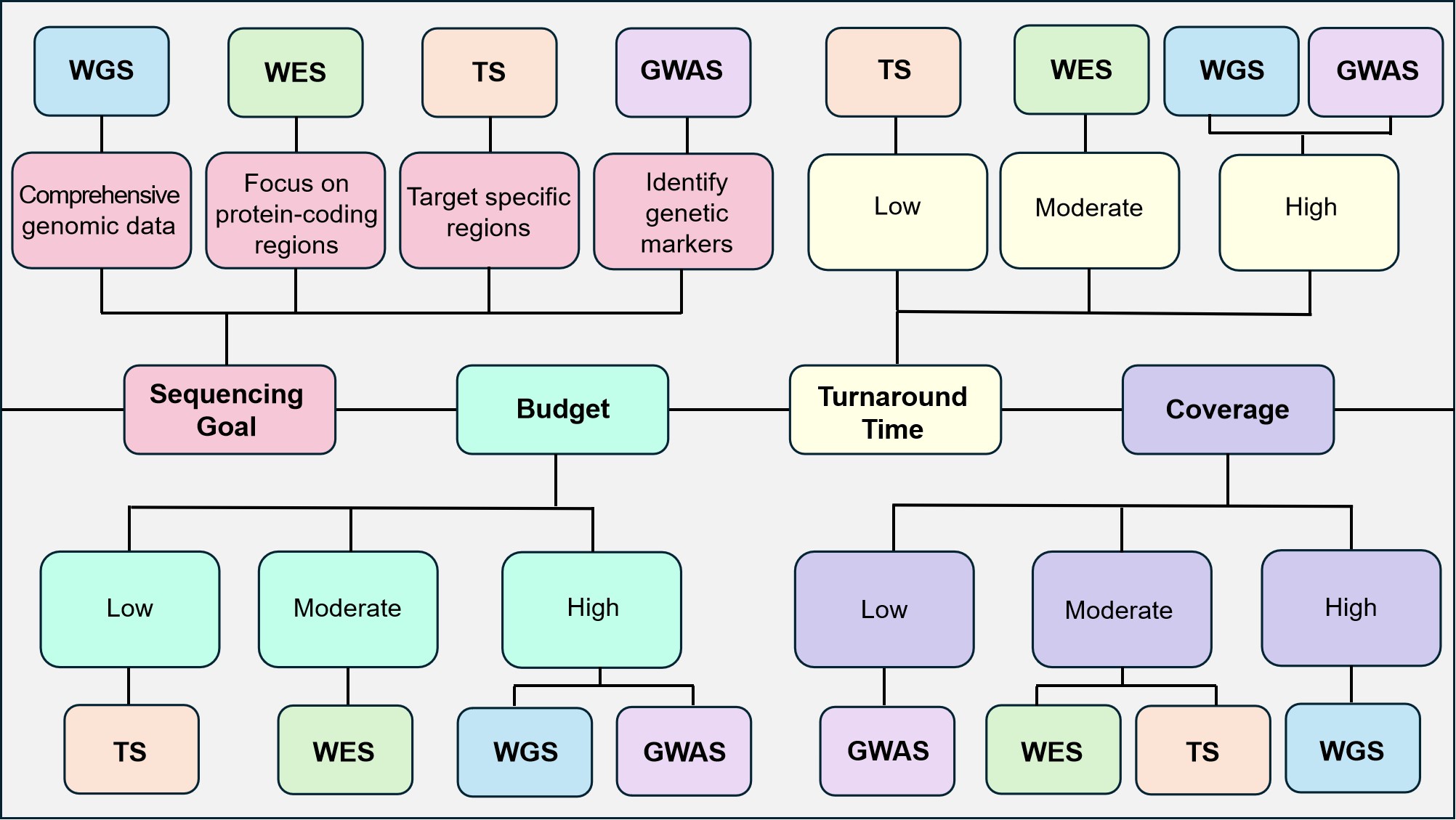
Figure 15. Choosing the Right Sequencing Method Is an Important First Step in Broadening Our Understanding of Neurological Disorders and Developing Better Diagnostics and Prognostics. Some key factors to be considered when making the choice include the end goal of the sequencing, how much funding is available, and if time and specific coverage are important requirements.
While methods like WGS, WES, TS, and GWAS provide powerful tools for analysing genetic data, they represent only part of the story. To truly comprehend how genetic variations interact with other biological factors, we need to look beyond genomics. Multi-omics approaches which can integrate genomics with additional biological data layers to provide a more thorough knowledge of disease causes and possible treatments will be discussed in the following section.
4 MULTI-OMICS APPROACHES
Upon examination of the several genomic techniques such as WGS, WES, TS, and GWAS, it is evident that each provides distinct perspectives on the genetic makeup of neurological illnesses. However, to truly understand these complex diseases, integrating data from beyond just the genome is key, and this is where multi-omics approaches come into play.
Multi-omics approaches provide a detailed understanding of disease mechanisms by combining data from different biological layers (Figure 16). By integrating multiple omics data, researchers can uncover novel genetic variants and disease genes that may not be apparent from single-omics studies[93,94].
|
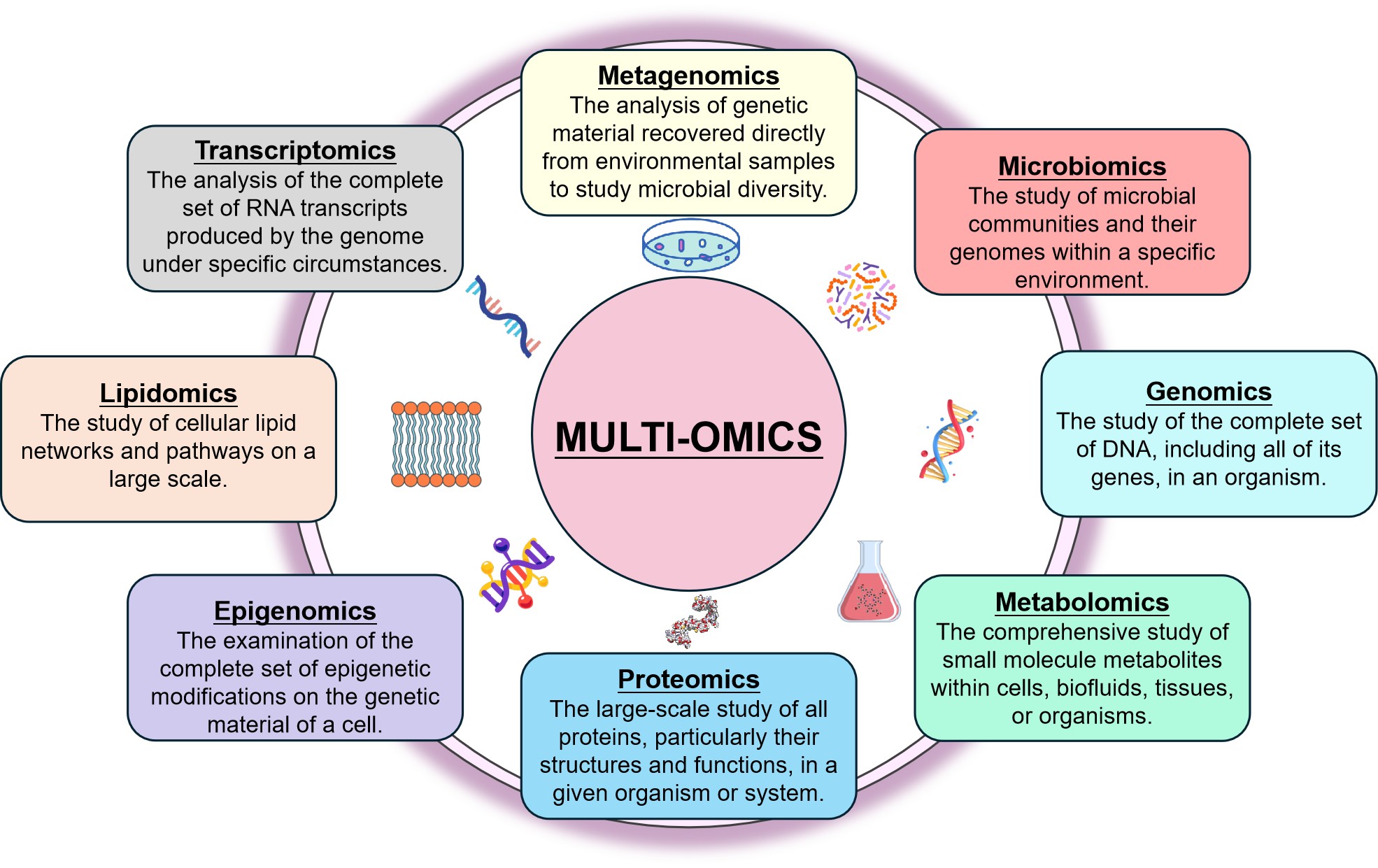
Figure 16. Multi-Omics Integration. Multi-omics combines fields like genomics, transcriptomics, proteomics, and metabolomics to provide a comprehensive understanding of biological systems, aiding in the discovery of disease mechanisms, therapeutic targets, and biomarkers.
The multi-omics approach has significantly enhanced our understanding of complex diseases such as AD, PD, and various psychiatric disorders, leading to the identification of new therapeutic targets and biomarkers for improved clinical outcomes[93,94]. Additionally, it has facilitated the discovery of shared genetic patterns and pathways across different neurodegenerative and neuropsychiatric conditions, offering deeper insights into their aetiology and progression[93,94]. OʼConnor et al.[95] emphasised the power of integrated transcriptomics and proteomics in identifying differentially expressed genes (DEGs) and proteins in diseases like MS, whilst Zhao et al.[96] showed how network-based approaches combining genomics, transcriptomics, and proteomics have identified key regulatory pathways and hub genes in AD.
4.1 Integration of Omics Data
A recent study by Le Grand et al.[97] used a combination of GWAS and transcriptome-wide association studies (TWAS) to investigates the genetic underpinnings of cerebral small vessel disease (cSVD), a major cause of stroke and dementia, using advanced diffusion imaging techniques (Figure 17). This multi-omics approach allowed for the integration of genetic data from GWAS with gene expression profiles from TWAS, enabling the identification of genes whose expression levels are influenced by genetic variants associated with cSVD. The study was able to connect certain genetic variations with their functional effects at the transcriptome level by merging these two levels of omics data, which led to a more thorough knowledge of the molecular processes driving cSVD. Single-omics research could have missed 32 genes whose expression was substantially correlated with NODDI indicators, but this multi-omics method found them. In particular, the combination of GWAS and TWAS data demonstrated that the expression of these genes in the brain and vascular organs suggests a potential involvement in early-life pathways leading to cSVD. This method also highlighted the concept of early treatment and prevention measures as they demonstrated that the white matter architecture is impacted by genetic predisposition to cSVD beginning in early life.
|
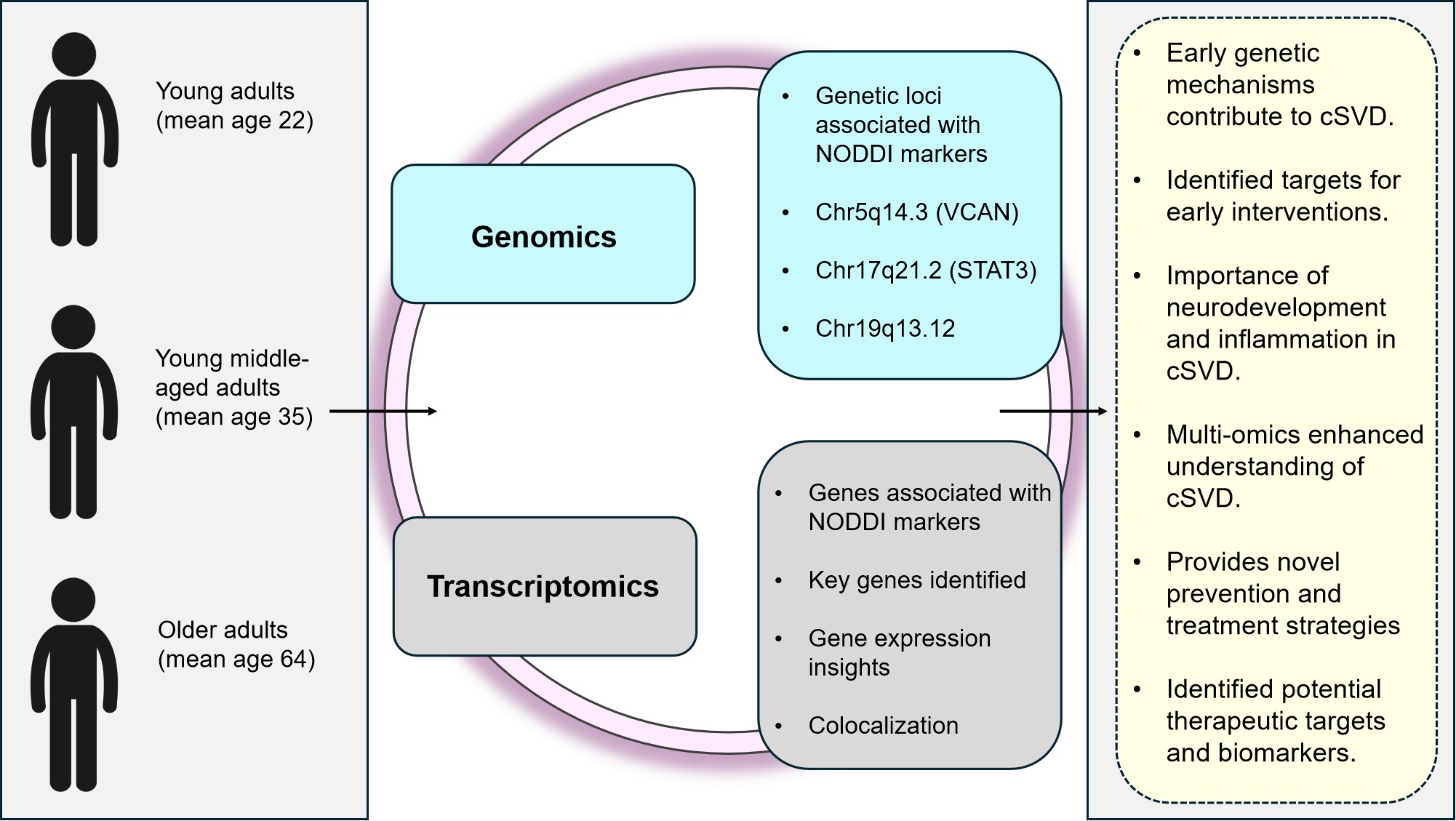
Figure 17. Summary of Study[97] on Cerebral Small Vessel Disease (cSVD). The figure illustrates participant age groups and key findings from a multi-omics approach. Genomic loci (e.g., Chr5q14.3, Chr17q21.2, Chr19q13.12) and transcriptomic data identify genes associated with NODDI markers. The study highlights early genetic mechanisms in cSVD, emphasizing neurodevelopment, inflammation, and potential therapeutic targets, suggesting that genetic influences on brain structure begin early in life.
In the study by Shaath et al.[98], a multi - omics analysis provided a detailed understanding of the disease mechanisms in monozygotic female twins with a rare neurodevelopmental disorder. These twins, born to related Iranian parents, exhibited a complex clinical profile including polymicrogyria, respiratory distress, and multi-organ dysfunction conditions that traditional diagnostics could not fully explain.
Table 5 summarises the clinical features observed in twins with a rare neurodevelopmental disorder, providing a foundation for the genetic and metabolic analyses that follow. It highlights key symptoms, such as polymicrogyria, respiratory distress, and multi-organ dysfunction.
Table 5. Summarises the Clinical Features Observed in Twins with a Rare Neurodevelopmental Disorder (Adapted from Ref.[98])
Clinical Feature |
Description |
Polymicrogyria |
Extensive polymicrogyria with frontoparietal predominance, patchy white matter changes, brain atrophy. |
Respiratory Distress |
Chronic respiratory issues observed in both twins. |
Multi-organ Dysfunction |
Dysfunction involving kidneys and heart. |
The integration of WGS with untargeted metabolomics was key to unravelling the underlying disease mechanisms. WGS identified two rare homozygous variants p.Arg565Trp in ADGRG1 and p.Glu910Val in CNTNAP1, genes known to be associated with polymicrogyria and hypomyelinating neuropathy. By combining genetic data with comprehensive metabolic profiling, the study was able to map how these variants influenced broader metabolic pathways, providing a more detailed and nuanced understanding of the disorder, especially in a consanguineous population. Table 6 details these genetic findings.
Table 6. Presents the Genetic Findings from WGS (Adapted from Ref.[98])
Gene |
Variant |
Associated Disorder |
ADGRG1 |
p.Arg565Trp |
Autosomal recessive polymicrogyria. |
CNTNAP1 |
p.Glu910Val |
Hypomyelinating neuropathy. |
Metabolomics analyses showed prominent disruptions in lipid and amino acid pathways related to oxidative stress, with metabolic changes correlating with the symptoms observed in the twins. These insights, listed in Table 7, into the physiology that genetics data alone could not show were essential to demonstrate the value of multi-omics to appreciate mechanisms of disease. This was further supported by the MRI scan, revealing brain abnormalities consistent with the phenotypic consequences of the genetic variants. This case illustrates the enhanced value of multi-omics approaches in detailing the intricate elements and processes underlying complex diseases, making them indispensable in identifying potential therapeutic targets. Table 8 comprises information on the multi-omics technologies used and critical observations from each omics level.
Table 7. Outlines the Metabolic Pathway Alterations Identified Through Metabolomic Analysis (Adapted from Ref.[98])
Metabolic Pathways |
Alteration |
Implication |
Lipid Metabolism |
Dysregulation in sphingolipids and phosphatidylcholines. |
Cellular damage and oxidative stress. |
Urea Cycle |
Accumulation of urea cycle metabolites. |
Potential defect in ammonia detoxification. |
Amino Acid Metabolism |
Elevated methionine sulfoxide and polyamines; depleted glutathione. |
Oxidative stress and antioxidant depletion. |
Table 8. Details the Multi-omics Approach Used by Ref.[98]
Omics Layer |
Technology Used |
Key Findings |
Genomics |
WGS |
Identified ADGRG1 and CNTNAP1 variants, elucidating the genetic basis of the disorder. |
Metabolomics |
Untargeted Metabolomics |
Revealed disruptions in lipid and amino acid metabolism, indicating oxidative stress. |
Perhaps the definitive example, to date, of a multi-omics approach to studying a neurodegenerative disorder comes from Van Karnebeek et al[99]. In this study, the researchers employed a highly integrative multi-dimensional approach, combining proteomics, metabolomics, lipidomics, genomics, computer modelling, clinical data assessment, and biochemical tests. This comprehensive strategy was designed to decipher the complex pathways through which specific genetic defects contribute to neurodegeneration. The cohorts under investigation included three patients with microcephaly, congenital brain abnormalities, progressive neurological impairments, recurrent infections, and ultimately fatal outcomes, who were referred for diagnostic investigation at the Amsterdam University Medical Centre, along with two control individuals (Figure 18).
The integration of omics data began with genomic DNA isolated from patient-derived fibroblasts undergoing whole exome sequencing (WES), which identified missense variants in the CIAO1 gene in two patients and a homozygous in-frame deletion in the MMS19 gene in the third patient (Figure 18). These genetic variants were absent in the controls and were predicted to disrupt protein function. Biochemical analysis revealed elevated quantities of uracil and thymine in the bodily fluids of the patients. To further elucidate the impact of these mutations, DPD enzyme activity assays were conducted in fibroblasts at varying temperatures.
|
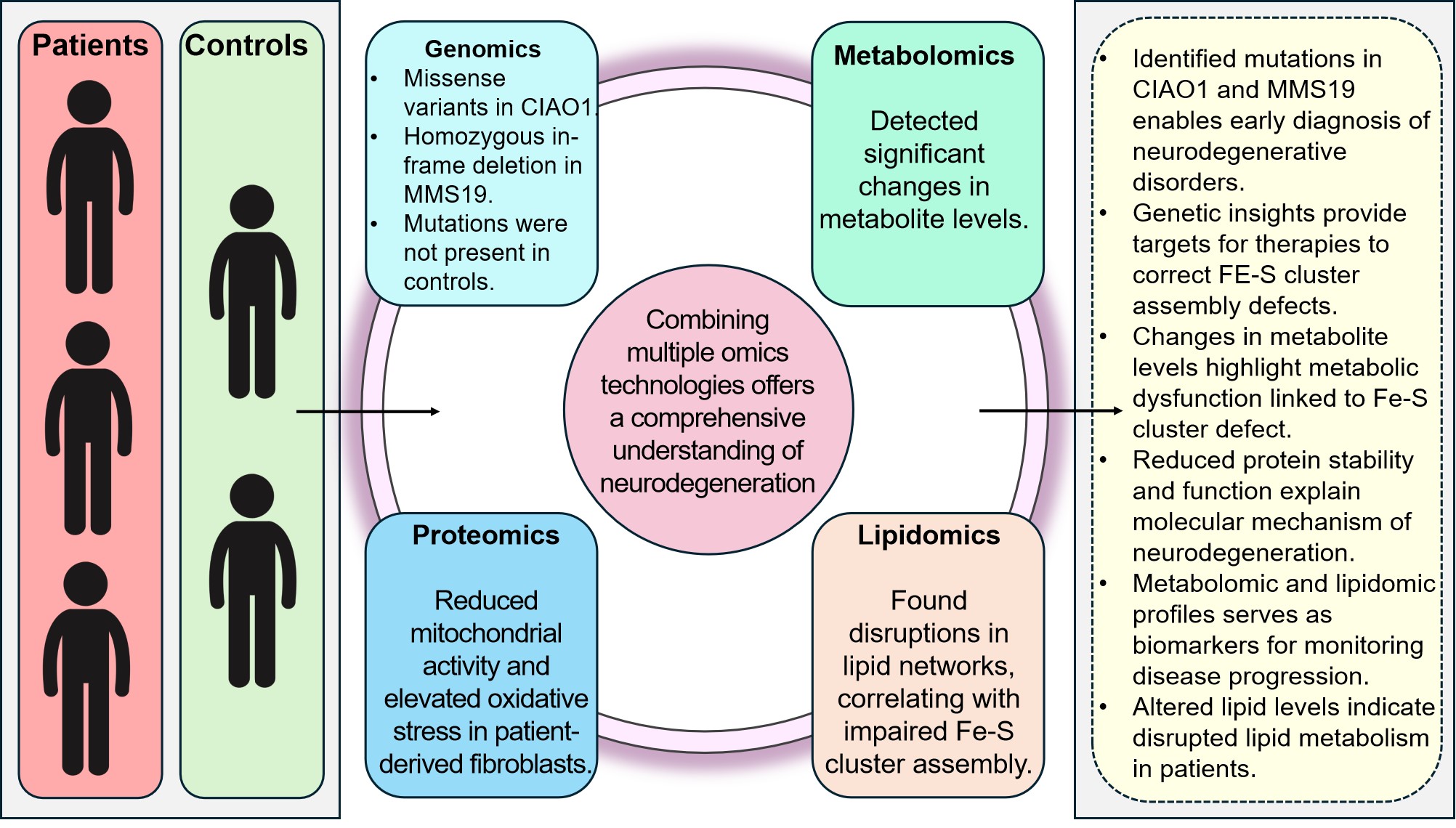
Figure 18. Summary of Multi-Omics Study[99] on Neurodegenerative Disorders. Highlights findings across genomics (mutations in CIAO1 and MMS19), proteomics (reduced mitochondrial activity, elevated oxidative stress), metabolomics (changes in metabolite levels), and lipidomics (disrupted lipid networks). By integrating these omics approaches, the study offers a comprehensive understanding of neurodegeneration linked to Fe-S cluster assembly defects, identifying potential biomarkers and therapeutic targets.
Mass spectrometry facilitated a detailed proteomic analysis, while ultra-high-pressure liquid chromatography (UHPLC) coupled with high-resolution mass spectrometry was employed for lipidomic analysis. High-performance liquid chromatography (HPLC) coupled with tandem mass spectrometry was utilised for metabolomic analysis. Each of these omics layers provided distinct insights, but their integration was critical in revealing how these mutations led to a cascade of cellular dysfunction.
In silico protein structure analysis was performed to predict the effects of the mutations on protein stability, predictions that were subsequently validated using CRISPR/Cas9-engineered homozygous zebrafish lines with loss-of-function alleles in ciao1 and mms19. These zebrafish exhibited neurodegenerative phenotypes similar to those observed in the patients. Functional assays confirmed that these mutations caused reduced protein stability and impaired Fe-S cluster assembly, evidenced by decreased DPD activity in patient fibroblasts. The multi-omics approach further revealed wide-ranging disturbances in cellular metabolism, including reduced mitochondrial activity and elevated oxidative stress, as demonstrated by extensive changes in the proteome, metabolome, and lipidome of patient-derived fibroblasts.
The study by Van Karnebeek et al.[99] illustrates that abnormalities in the CIAO1 and MMS19 proteins result in profound metabolic disruptions and defective Fe-S protein maturation, ultimately leading to a fatal neurodegenerative disease. The results from this exhaustive multi-omics approach have identified novel therapeutic targets related to Fe-S cluster formation disruption that, hitherto, had remained unknown. In addition, it has provided some fresh information on the molecular causes of neurodegeneration.
Integrating data from genomics, proteomics, metabolomics, and other fields has given way to the identification of new biomarkers, discovery of disease mechanisms, and even the suggestion of potential therapeutic targets not detectable with genomic data alone. Although multi-omics has made colossal progress in the field, it has essentially been applied to the more common conditions. The unique challenges of rare neurological disorders very often require even more tailored approaches.
We review how multi-omics is being used to treat rare neurological disorders in the following section.
4.2 Omic Investigation of Rare Neurological Disorders
It is in the area of the more common neurological conditions that genomic approaches, especially WGS, WES, and GWAS, have contributed much. A more holistic approach may be needed, however, for rare neurological disorders. Multi-omics data related to genomics, proteomics, metabolomics, and other disciplines can shed important insights into intricate biological networks driving rare conditions.
With so few effective therapies available for sporadic Creutzfeldt-Jakob disease, Jiang et al.[100] aimed to identify genetically-supported treatment targets as a means to combat this rapidly progressive and fatal neurodegenerative condition. Utilising a comprehensive multi-omics approach with data from 13,569 controls and 4,110 sporadic creutzfeldt-jakob disease (sCJD) patients, predominantly of European descent, the study integrated TWAS, proteome-wide association studies (PWAS), and epigenome-wide association studies (EWAS). More accurate identification of possible therapeutic targets was made possible by this integration, which single-omics approaches may not have been able to provide. Using information from research cohorts like GTEx and ROSMAP, the study further highlighted cis-expression QTLs (cis-eQTLs), single-cell expression QTLs (cis-sc-eQTLs), DNA methylation QTLs (cis-mQTLs), and protein QTLs (cis-pQTLs) in blood and brain tissues.
Utilising Bayesian colocalisation analyses and Mendelian randomisation (MR), which were essential for showing causal relationships between genetic variants and sCJD, demonstrates the studyʼs statistical soundness. In order to increase the likelihood that the relationships revealed are indeed causal, these techniques help reduce confounding variables and reverse causality. MR analysis leverages genetic variants as instrumental variables to infer causality, and Bayesian colocalisation further validates these findings by assessing whether the same genetic variants influence both the exposure (e.g., gene expression) and the outcome (sCJD risk). The research identified 23 potential therapeutic targets for sCJD, with five key genes standing out: STX6, XYLT2, PDIA4, FUCA2, and KIAA1614. Notably, STX6 emerged as the highest-ranked target, with its expression in neurons and oligodendrocytes being linked to an increased risk of sCJD in brain regions such as the cortex and striatum, which are commonly affected by the disease.
Through the integration of multi-omics data such as TWAS, PWAS, and EWAS differential gene expression across several brain areas and cell types was discovered, providing deeper insights into the fundamental disease mechanisms of sCJD. The various brain areas where STX6, FUCA2, and KIAA1614 are expressed demonstrate the intricacy of sCJD aetiology and point to potential novel targets for therapy that may have gone unnoticed in less thorough techniques.
Although limited by the lack of any other sCJD GWAS data and a predominantly European study population, this study provided some solid background for further research. In order to investigate the biological mechanisms behind these targets and carry out clinical trials to assess prospective therapies for sCJD, it is necessary to identify viable therapeutic targets and consider the possible repurposing of currently available medications through multi-omic integration.
Table 9 details the omics technologies utilised in the study, along with the findings, potential applications, and additional insights derived from each omics layer. The identification of key genes such as STX6, XYLT2, PDIA4, FUCA2, and KIAA1614 highlights the studyʼs contribution to understanding the pathophysiology of sCJD and the identification of promising drug targets.
Table 9. Integrative Multi - Omics Analysis in Identifying sCJD Therapeutic Targets (Adapted from Ref.[100])
Omics Technology |
Findings |
Potential Applications |
Additional Insights |
Transcriptomics |
Key Genes Identified: STX6, XYLT2, PDIA4, FUCA2, KIAA1614. |
Therapeutic Targets: Identified potential targets like STX6 for drug development. |
STX6 and XYLT2 show high expression in neurons and oligodendrocytes. Differential expression patterns observed in cortex and striatum. High expression of STX6 associated with increased sCJD risk. |
Differential expression patterns observed in cortex and striatum. |
Gene-Disease Associations: TWAS provided insights into gene-disease links. |
||
Proteomics |
Altered protein levels linked to sCJD, including PDIA4 and FUCA2. |
Biomarker Discovery: Potential biomarkers for diagnosis and prognosis. |
Proteins identified in specific brain regions and their association with sCJD. Supports understanding of disease mechanisms at the protein level. |
PWAS conducted. |
Therapeutic Interventions: Targeting protein expression and function. |
||
Epigenomics |
DNA methylation patterns linked to sCJD risk. |
Epigenetic Therapies: Targeting methylation changes to influence risk. |
EWAS identified key methylation sites. Provides additional regulatory insights over genetic findings. |
Analysis of cis-mQTLs in blood and brain tissues. |
Regulatory Insights: Understanding how epigenetic changes affect gene expression. |
||
Multi-Omics Integration |
Combined omics analyses revealed 23 potential therapeutic targets. |
Holistic Approach: Combines multiple omics for a comprehensive view. |
STX6 consistently identified as a high-priority target across studies. XYLT2 and FUCA2ʼs involvement supports drug repurposing. |
Mendelian randomization and Bayesian colocalization used for causal insights. |
Drug Repurposing: Identified interactions with existing drugs like carboplatin. |
MECP2 duplication syndrome (MDS) is a rare X-linked neurodevelopmental disorder that primarily affects men and is caused by the duplication of the MECP2 gene and at least one other gene, often IRAK1, located on the Xq28 region of the X chromosome. The MECP2 protein, which is essential for healthy brain development and function, is overexpressed as a result of this duplication. Because of X chromosomal inactivation, females are often asymptomatic carriers of the condition, though some may exhibit symptoms.
The study by Pascual-Alonso et al.[101] examines MDS through transcriptomics and proteomics to uncover altered pathways and potential therapeutic targets. By integrating these omics layers, the researchers linked mRNA changes with protein modifications, identifying key pathways related to immune system control, vesicular transport, and cytoskeletal function. For instance, the downregulation of KIF3B in both analyses highlighted its role in impaired vesicular transport in MDS. The study also compares MDS with Rett Syndrome (RTT), noting that MDS results from gene duplication while RTT involves loss-of-function mutations.
The study involved 61 participants: 17 MDS patients (15 males, 2 females), 10 asymptomatic carriers, 21 RTT patients, and 13 controls. Using skin fibroblast cell lines, transcriptome analysis was performed with NextSeq 500 and DESeq2, while proteomics used MaxQuant and TMT-mass spectrometry, with Limma in R for analysis. Enrichment analysis, using clusterProfiler and ReactomePA, focused on Gene Ontology and KEGG pathways with a p-value threshold of 0.05, corrected using the Benjamini-Hochberg method.
The findings revealed 2,465 DEGs and 300 differentially expressed proteins (DEPs) in male MDS patients, with key dysregulations in the cytoskeleton, synapse shape, and cell migration. Despite classifying patients by duplication size and position, no significant genotype-phenotype associations were found. In female MDS patients, 5,720 DEGs and 493 DEPs were identified, particularly affecting translation processes and splicing, with three genes (ABCC4, STK17B, MYO1C) dysregulated in both genders.
Carriers showed 2,888 DEGs and 635 DEPs, with minimal dysregulation in cell cycle and splicing, possibly explaining their asymptomatic nature. Comparing MDS and RTT revealed 721 shared DEGs and 12 DEPs, highlighting distinct pathways: MDS showed enrichment in mRNA processing, while RTT was enriched in cell adhesion pathways.
Numerous biomarkers and therapeutic targets were identified, including TMOD2, SRGAP1, and KIF3B. The study emphasized the need for larger cohorts, especially of female patients, and the integration of patient-derived samples and neuronal models to advance therapeutic strategies for MDS.
And Figure 19 highlights the number of DEGs and proteins (DEPs) across various cohorts, including Male MDS, Female MDS, Carriers, and RTT. Figure 20 provides a conceptual illustration of pathway enrichment scores, comparing the contributions from transcriptomics, proteomics, and their combination, with values intended to emphasise the varying impacts of these omics technologies.
|
Figure 19. The Number of DEGs Identified Through Transcriptomics and Differentially Expressed Proteins (DEPs) Identified Through Proteomics Across Different Cohorts (Male MDS, Female MDS, Carriers, RTT).
|
Figure 20. The Enrichment Scores of Various Pathways, Comparing the Contributions from Transcriptomics, Proteomics, and Their Combination. The values shown are illustrative and not derived from specific study data, intended to highlight how different omics technologies may contribute to pathway enrichment.
A recent study by Segarra-Casas et al.[102] explored a female patient presenting with Duchenne muscular dystrophy (DMD)-like symptoms, including frequent falls, calf hypertrophy, and muscle weakness starting at age 7. Despite these symptoms, the patient remained undiagnosed for over two decades, as standard genetic tests such as multiplex ligation-dependent probe amplification (MLPA) and exome sequencing failed to identify the underlying cause. DMD is a severe form of dystrophinopathy caused by mutations in the DMD gene, which encodes the dystrophin protein essential for muscle fiber integrity. While DMD predominantly affects males, a small percentage of female carriers can exhibit symptoms due to factors such as skewed X-chromosome inactivation.
This work demonstrated the effectiveness of multi-omics techniques in detecting intricate genetic changes through the application of WGS and RNA sequencing (RNAseq). RNAseq provided a global view of gene expression and revealed an 85% reduction in DMD gene expression compared to 116 muscle samples in the cohort (P=1.6×10-11). This statistically significant reduction indicated a major disruption in gene function. WGS further allowed for the precise identification of a de novo balanced translocation between chromosome 17 and the X chromosome. A more thorough knowledge was obtained by combining transcriptomics and genomics, while traditional single-omics approaches were unable to pinpoint the genetic abnormality underlying the patientʼs condition.
The experimental approach included rigorous validation techniques. Allele-specific PCR and karyotyping were used to confirm the translocation, and the disruption of the Dp427 muscle isoform of the DMD gene was confirmed, providing a clear genetic explanation for the patient’s symptoms. By analysing RNAseq data using statistical methods like the detection of RNA outliers pipeline, the researchers were able to determine that DMD was the only outlier gene in the patientʼs sample. The dependability of the results is shown by the combination of statistical validation and experimental methodologies.
This case study illustrates how complicated structural variations in female carriers can lead to disease manifestations. The patientʼs manifestation of a DMD - like phenotype as a result of the DMD gene disruption emphasises the necessity of extensive testing above and beyond recommended guidelines. The case also illustrates the difficulty traditional genetic testing faces in detecting complex chromosomal rearrangements. In addition to providing a useful diagnostic tool for neuromuscular disorders, the work of Segarra-Casas et al.[102] demonstrates the value of RNAseq and WGS in diagnosing uncommon genetic diseases and overcoming diagnostic obstacles (Table 10).
Table 10. Summary of Analytical Techniques and Their Diagnostic Implications in a DMD-like Case
Diagnostic Methods |
Findings |
Implications |
RNAseq |
85% reduction in DMD gene expression compared to the control cohort. Detected absence of reads from exon 14 to 62, indicating a large deletion or structural variant. |
Illustrates the effectiveness of RNAseq in detecting gene expression abnormalities that standard tests may miss. Helps prioritize WGS analysis. |
WGS |
Identified a de novo balanced translocation between chromosome 17 and the X chromosome (t(X;17)(p21.1;q23.2)). Disruption of the DMD gene on the X chromosome and BCAS3 gene on chromosome 17. |
Highlights the limitations of traditional genetic testing in identifying complex chromosomal rearrangements. Provides a conclusive diagnosis for the patientʼs symptoms that were undiagnosed for over two decades. |
Histopathological Analysis |
Confirmed dystrophic features in muscle biopsies, including fiber size variability, internal nuclei, and significant adipose tissue replacement. Significant reduction in dystrophin expression. |
Supports the genetic findings by linking structural variants to observable muscle pathology. Confirms the muscle tissueʼs abnormal structure, consistent with dystrophinopathy. |
Immunohistochemistry & Western Blotting |
Diminished staining of dystrophin and associated proteins (e.g., α, β, γ sarcoglycans and β-dystroglycan). Faint detection of dystrophin rod domain by Western blot. |
Validates the reduction of dystrophin and associated protein levels, reinforcing the genetic diagnosis and showing severe dystrophic impact on muscle fibers. |
Notes: An overview of the diagnostic findings and implications in a case of Duchenne Muscular Dystrophy-like symptoms, highlighting the effectiveness of RNA sequencing and whole genome sequencing in providing a conclusive genetic diagnosis.
Integrating diverse biological data has been made possible by the use of multi-omics in rare neurological disorders. Figure 21 provides a visual comparison of key metrics across the three studies: sCJD, DMD, and MDS. It highlights the different focuses and outcomes of each study, including the use of omics techniques, identification of therapeutic targets, and diagnostic achievements.
|
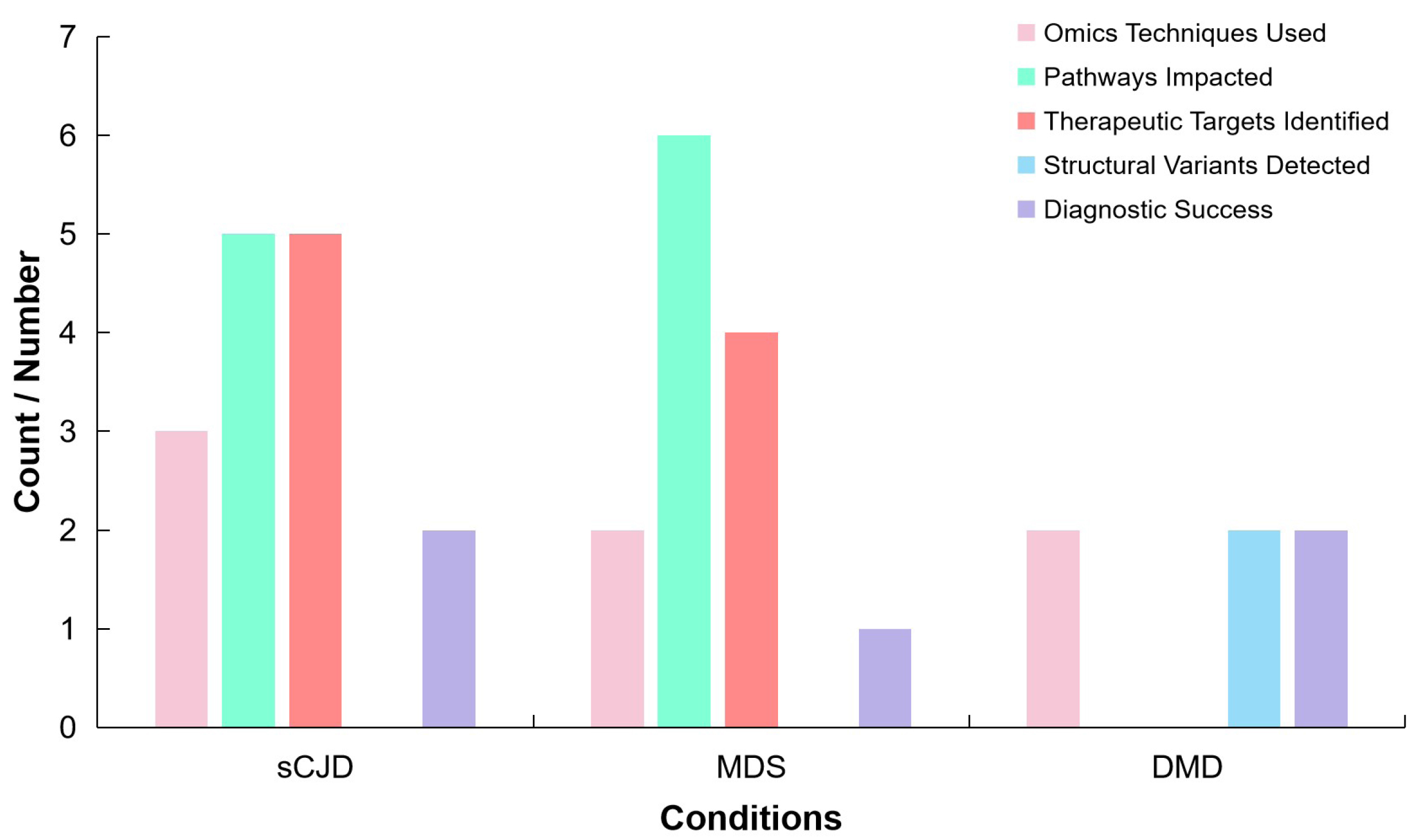
Figure 21. The Relative Focus and Outcomes Across Three Studies on sCJD, MDS, and DMD. The chart highlights the use of omics techniques (pink), the identification of therapeutic targets (coral), the impact on biological pathways (light green), the detection of structural variants (light blue), and diagnostic success (purple) in each study. While the chart does not represent exact numerical data, it provides a visual summary of the key areas emphasized by each study based on qualitative analysis. The categories reflect general trends and findings, offering a comparative overview of the research efforts and their implications in these diseases.
The genetic variety that occurs across populations worldwide must also be taken into account if we are to fully exploit the advantages of these strategies. Not doing so could restrict how broadly applicable our findings are and result in under-representation of particular populations in studies and medical interventions. In order to demonstrate how inclusive research may result in more fair and efficient healthcare outcomes, the value of diversity in genomics will be discussed in the next section.
5 LACK OF DIVERSITY IN GENOMIC STUDIES
To this point, we have discussed how genomic research has helped advance our understanding or neurological disorders, and their potential treatments. One of the main challenges is the scarcity of genomic diversity within these studies. This section will consider the consequences of this underrepresentation on the generalisability from genomic studies of neurological disorders and discuss associated limitations in developing treatments that are at one time inclusive yet effective.
5.1 Population Bias
The underrepresentation of non-European populations in genomic research continues to limit our global understanding of neurological disorders[73], particularly in rarer diseases[50] and genetic risk factors[92,103]. Jonson et al.[103] highlight that 82% of studies on Alzheimer’s and Parkinson’s involve predominantly European participants, leaving non-European cohorts significantly underrepresented, especially in conditions like Lewy body dementia and frontotemporal dementia. This lack of diversity hinders the development of targeted therapies, underscoring the need for more inclusive research to identify genetic variants across diverse populations.
Similarly, Rutten-Jacobs et al.[104] examined racial and ethnic diversity in neuroscience clinical trials and found that White participants were overrepresented (85.6%), while Black (1.6%) and Hispanic / Latino (13.7%) participants were underrepresented. Their analysis points to the need for broader recruitment strategies, community involvement, and digital tools to enhance diversity in clinical trials.
5.2 Access to Resources
Khani et al.[105] emphasise the critical role of inclusive research in understanding the genetic diversity associated with PD. Historically, PD genetics research has centred on European populations, leading to missed opportunities for early diagnosis and tailored treatments in non - European groups. Their work identifies novel genetic loci associated with PD, such as GBA1 in Africans and STXBP6 in Latinos, underscoring the need for diversified models of research. In comparison to previous large-scale studies of Parkinsonʼs genetics, GP2 adds samples from a greater range of ethnic backgrounds into the Global Parkinsonʼs Genetics Programme, which better allows it to derive treatments that are more precise and generalisable.
Diverse and inclusive research is necessary to completely understand the spectrum of neurological disorders. The complexity of neurological illnesses and the ways in which different variables influence their onset and course will be discussed in more detail in the next section.
6 COMPLEXITY OF NEUROLOGICAL DISORDERS
Although reducing inequality in genomics research is very important to ensure quality healthcare delivery, it points toward the intrinsic difficulty of neurological disorders. As discussed in previously, neurological disorders present a wide array of clinical symptoms and underlying genetic factors. These illnesses are extremely complicated due to their polygenic origin and complex interactions between genes and environmental variables. This section explores these nuances in more detail, emphasising how they impede our understanding and treatment of neurological disorders, and the difficulties presented by alternate diagnosis and therapy options.
6.1 Polygenic Nature
Neurological illnesses often involve multiple genes, making them polygenic and complex to understand. Tanaka and Vécsei[106] highlight this in disorders like epilepsy and febrile seizures, where SCN1A mutations interact with other genes, contributing to the condition. Similarly, Zhang et al.[11] illustrate that familial hemiplegic migraine (FHM) involves not only ATP1A2 mutations but also other genes like CACNA1A and SCN1A, which collectively affect neuronal function. This interplay complicates the mapping of specific genes in polygenic diseases. Mavroudis et al.[107] extend this to functional neurological disorders (FNDs), where subtle genetic abnormalities increase vulnerability, requiring other factors to trigger symptoms. Caznok Silveira et al.[108] argue that complex brain network disruptions in conditions like autism and schizophrenia arise from multiple interacting genetic factors, underscoring the need for a comprehensive approach to understanding these disorders.
6.2 Gene-Environment Interactions
The interaction between gene and environment add another layer of complexity to neurological disorders. Tanaka and Vécsei[106] discussed how in ADHD, genetic predispositions related to dopamine regulation interact with environmental factors like stress or diet, influencing the disorder’s severity. Mavroudis et al.[107] explore how stress and trauma interact with genetic vulnerabilities in FNDs, where environmental triggers often initiate symptoms. Mayston et al.[109] take a systems science perspective on neurodevelopmental disorders like cerebral palsy, highlighting how prenatal environmental factors intertwine with genetic predispositions. Tripathi et al.[110] further explain that gene-environment interactions, such as the interaction between the APOE ε4 allele and environmental factors like pollution, significantly impact dementia risk. These cases demonstrate the need for a holistic approach to treating neurological diseases, considering both genetic and environmental factors throughout a patient’s life.
Part of what makes neurological disorders so complex is the array of factors interconnected and interwoven that drive their underlying mechanisms, hence, hard to fully decipher. But complexity is not the only challenge. The limitations of present technology, ethical considerations, and the need for far more specific therapies if the area is to advance are just a few of the obstacles that will be discussed in the following section.
7 ADDITIONAL CHALLENGES IN GENOMIC RESEARCH
A number of other difficulties that can impact the efficacy and applications of genomic research exist in addition to the particular problems of diversity and complexity that were previously highlighted. Additional difficulties that will significantly influence the future course of genetic research into neurological illnesses are discussed in this section. The obstacles stem from ethical quandaries, issues with data processing, and constraints with technology.
7.1 Ethical Considerations
7.1.1 Privacy and Confidentiality
The sensitivity of genomic data, especially with the rise of personalised medicine, presents significant privacy and confidentiality challenges[111-113]. Advances in NGS and broader data access increase the risk of breaches unless strict data-sharing protocols and robust security measures are in place. Handling genomic data also requires culturally sensitive approaches to ensure privacy expectations are respected across diverse populations[111].
7.1.2 Informed Consent
The complexity of genetic data makes informed consent challenging. As whole genome sequencing becomes more common, patients often struggle to grasp the full implications of their data usage, particularly regarding future reanalysis[112,114]. Clinicians also face difficulties in communicating uncertainties in prognoses, especially in severe neurological cases[115]. These challenges highlight the need for clear communication and robust consent processes in the evolving field of genomics[112,114,115].
7.1.3 Genetic Discrimination
Genetic information can lead to stigmatisation and discrimination, particularly in communities where hereditary conditions are prevalent[111,116]. This issue is further emphasised in the UNESCO International Declaration on Human Genetic Data, which specifically addresses the ethical necessity of preventing discrimination and stigmatisation based on genetic information. As genetic data becomes increasingly utilised in medical applications, there is a growing risk of misuse by employers, insurers, or other entities. Addressing this issue requires ongoing vigilance and strong ethical frameworks[115,117].
These points collectively highlight the ethical and practical challenges of ensuring that genetic information is handled in a way that prevents discrimination and stigmatisation, particularly as its use in various sectors continues to expand.
7.2 Technological and Methodological Challenges
7.2.1 Data Analysis and Interpretation
The vast amount of data generated by NGS poses challenges in interpretation and analysis, requiring advanced computational methods and a skilled workforce[112,113]. As genomic knowledge evolves, systematic reanalysis of data can improve diagnostic accuracy but also raises ethical concerns about privacy and consent[112,117,118]. Effective communication of complex genetic information to the public and translation into local languages is crucial for ethical research and community engagement[118].
7.2.2 Reproducibility and Validity
Variability in diagnostic outcomes due to differences in methods and data quality highlights the need for standardised protocols[12]. Ensuring validity across diverse populations is essential, particularly for complex disorders[109,110,112,113]. Cultural beliefs and participant retention also impact research consistency, necessitating tailored community engagement strategies and fair negotiations regarding research incentives[110,118].
7.3 Translational Challenges
7.3.1 Bridging the Gap to Clinical Practice
Translating research into clinical practice remains challenging, with a need for a cohesive, patient-centred research strategy that aligns with therapeutic needs[113,117,119]. Conditions like Rett syndrome and Duchenne Muscular Dystrophy illustrate the difficulties in matching scientific discoveries with practical clinical applications[11,12,102]. Bridging this gap requires ongoing efforts in translational bioinformatics and a focus on integrating complex data into actionable treatments[113,117].
7.3.2 Regulatory Hurdles
Regulatory challenges, particularly in personalised medicine, pose barriers to translating research into practice[113,119]. Harmonising policies across regions and ensuring robust frameworks for data security and privacy are essential[113,119]. As genomic technologies rapidly evolve, these frameworks must adapt to address emerging ethical dilemmas and support the implementation of new treatments[114].
7.4 Economic and Societal Considerations
The high costs of genomic sequencing and the potential for public distrust, particularly in public-private partnerships, pose economic and societal challenges[113,116,120]. Financial barriers and ethical concerns must be addressed to ensure sustainable implementation of genomic research[112]. Engaging communities and respecting cultural sensitivities are crucial for building trust and overcoming societal barriers to participation in genomic research[111,116].
The challenges we have reviewed in technical, ethical, and logistical dimensions are significant, but they also bring about the potential for innovation and growth. As we come to the end of this review, we will turn our thoughts to future prospects of neurological disorder research, with attention to how new technologies and collaborative efforts can break down such barriers to achieve better patient outcomes.
8 CONCLUSION AND FUTURE PROSPECTS
Neurological conditions can significantly impact an individualʼs quality of life[1]. As explained herein, a better understanding of these complicated conditions has been made possible by recent noteworthy advancements in the field of genomics, notably the clinical use of NGS technology. Thanks to the identification of significant variations in disease pathology markers made possible by advanced sequencing technologies like WGS, WES, TS, and GWAS, we have been able to develop superior therapeutic interventions and better diagnostic methods that have the potential to greatly improve patient health and well-being[121].
While WES focuses on the regions that code for proteins and has shown to be a potent clinical technique in the identification of rare genetic variants linked to neurological disorders[122], WGS offers a comprehensive overview of an individual’s full genetic complement, enabling the detection of numerous genetic variants[123]. WGS and WES provide valuable clinical insights[124] but come with challenges, including large data sets and the complex analyses requiring bioinformatics expertise[125]. TS, by comparison, is both a cost- and time-effective alternative, focusing only on the specific regions of interest that need to be sequenced[126]. GWAS further contribute to our understanding of neurological disorders by exploring common genetic variations across large cohorts[87]. This can help to refine phenotypes and improve the development of targeted therapies.
Nevertheless, despite its obvious importance as a powerful diagnostic and prognostic tool, genomics remains just one component in systems biology’s extensive toolbox of multi-omics technologies[127-129]. The ultimate goal of achieving a complete understanding of neurological disorders including all the complex interactions that exist at the genetic, epigenetic, and environmental levels[130] will likely only be fully achieved by a collective multi-omics approach (combining genomics with epigenomics, transcriptomics, proteomics, metabolomics, and more).
In this regard, multi-omics approaches are already doing very well in some complex diseases like Alzheimerʼs and Parkinsonʼs. Further studies should be conducted on the optimisation of such methods for rare neurological disorders, where the genetic causes have not been established in the majority of cases. Further research will be necessary for new biomarkers and therapeutic targets by combining genomics with other omics technologies, such as metabolomics and lipidomics. In addition, applying these approaches to paediatric neurology will significantly improve patient outcomes where the diagnosis and early intervention play a major role in such neurological disorders.
The future of this field is expected to focus on the integration of various multi-omics methods. By combining genomics with other omics layers[93-95], researchers can gain a deeper understanding of disease mechanisms, discover new biomarkers, and develop more personalised treatment plans. This integration has already shown promise in diseases such as Alzheimer’s and Parkinson’s, where combining genomic data with transcriptomics and proteomics has led to the identification of key regulatory pathways and potential therapeutic targets[96,97].
Challenges still remain, however. In clinical practice, one major drawback is the scalability of multi-omics technology. Applying these approaches on a large scale is difficult due to the high costs, complex data analysis, and the need for specialised bioinformatics tools. Moreover, although technologies can generate a huge amount of data, interpretation of the information within it remains a problem. Advanced computational tools and standardised frameworks are needed to find actionable clinical insights. More importantly, data-sharing agreements and standardisation across research institutions are essential to overcoming these challenges.
Apart from these difficulties, it is essential to ensure that diverse populations are included in genomic and multi-omics research. The under-representation of some communities in past genomic studies has limited the generalisability of the results to other ethnic groups. Future research should concentrate on inclusion and equity, ensuring that all communities benefit from the strides made in precision medicine. Another critical ethical concern involves inequities in access to such advanced technologies. Although precision medicine is being increasingly implemented in clinical practice, routes must be found that allow every patient, not just those of a certain socio-economic background, access to these new treatments.
This review has, therefore, emphasised that more research is needed in an attempt to establish just how multifaceted neurological disorders are particularly those that are rare or for which no definite diagnosis or management plans exist. In these efforts, genomics and multi - omics approaches have proven to be important tools. Only through the integration of these different data sets can we gain an appreciation of the subtlety of these illnesses, which is necessary for improving diagnostic techniques and therapeutic interventions. This review points to the need for continued effort in this direction and makes a case for thoroughness in its approach to these difficult disorders.
Utilising machine learning and artificial intelligence to examine the enormous amounts of data produced by these multi - omics techniques is one of the most promising future developments. Artificial intelligence-powered technologies can identify trends and connections that conventional analytic techniques might miss, leading to discoveries about disease causes and potential treatments[47]. Additionally, advancements in CRISPR and gene-editing technologies hold the potential to correct genetic mutations associated with neurological disorders[99].
Furthermore, it is anticipated that multi-omics data will be increasingly widely incorporated into clinical practice, enabling the creation of precision medicine strategies catered to the genetic profile of each individual patient. For individuals with neurological disorders, especially those with uncommon or challenging-to-treat problems, this might greatly improve outcomes[98,99,102].
Although genomics and multi-omics approaches have considerably advanced our understanding of neurological disorders, challenges still remain. The genetic basis remains poorly understood in most diseases, often due to the complexity of gene-environment interactions. Further investigations must prioritise the development of more accessible and accurate sequencing technologies, along with improvements in bioinformatics tools for better data interpretation. Therefore, to translate these research findings into clinical applications - turning multi - omics data into personalised treatment plans - collaborative efforts will be required across disciplines.
Accompanying these developments will be serious challenges: the establishment of sound data-sharing frameworks, standardised methodologies, and interdisciplinarity among researchers, clinicians, and bioinformaticians. Furthermore, ethical considerations, particularly concerning data privacy and the potential for incidental findings, must be carefully managed as we move towards an era of personalised medicine[64].
Though many important strides have been made in understanding and treating neurological disorders using genomics, the future clearly lies in coupling genomics to other omics technologies and applying advanced data analysis tools. It is therefore anticipated that further research and collaboration will be the formation, through these advancements, of a future in which diagnosis and treatment of neurological disorders shall be changed for the better and quality of life be enhanced for patients.
Acknowledgements
The authors would like to acknowledge the support of Munster Technological University, Cork, for providing the resources and environment that facilitated this work. TQ completed this work as part of an MSc in Computational Biology (https://www.mtu.ie/courses/crscobi9/) at MTU.
Conflicts of Interest
The authors declared no conflict of interest.
Data Availability
No additional data are available.
Copyright Permissions
Copyright © 2024 The Author(s). Published by Innovation Forever Publishing Group Limited. This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.
Author Contribution
Quirke T was responsible for conceptualisation, literature review, synthesis of findings, original draft writing, revisions, and creation of all visualisations and figures. Sleator RD was responsible for review and editing of the manuscript.
Abbreviation List
CMT, Charcot-Marie-Tooth disease
CNV, Copy number variation
DEG, Differentially expressed genes
DEP, Differentially expressed proteins
EWAS, Epigenome-wide association study
FHM2, Familial hemiplegic migraine type 2
GWAS, Genome-wide association studies
IPN, Inherited peripheral neuropathy
MDS, MECP2 duplication syndrome
MLPA, Multiplex ligation-dependent probe amplification
mQTL, Methylation quantitative trait loci
MS, Multiple sclerosis
NGS, Next-generation sequencing
ONT, Oxford nanopore technology
PCR, Polymerase chain reaction
PD, Parkinson’s disease
PWAS, Proteome-wide association study
RNAseq, RNA sequencing
RTT, Rett syndrome
sCJD, Sporadic creutzfeldt-Jakob disease
SMRT, Single molecule real-time sequencing
SNP, Single nucleotide polymorphism
TS, Targeted sequencing
TWAS, Transcriptome-wide association study
UMAP, Uniform manifold approximation and projection
VUS, Variants of uncertain significance
WES, Whole-exome sequencing
WGS, Whole-genome sequencing
References
[1] Feigin VL, Vos T, Nichols E et al. The global burden of neurological disorders: translating evidence into policy. Lancet Neurol, 2020; 19: 255-265.[DOI]
[2] Scheltens P, De Strooper B, Kivipelto M et al. Alzheimerʼs disease. Lancet, 2021; 397: 1577-1590.[DOI]
[3] Bloem BR, Okun MS, Klein C. Parkinsonʼs disease. Lancet, 2021; 397: 2284-2303.[DOI]
[4] Jen JC. Familial hemiplegic migraine. University of Washington Press: Seattle, USA, 2021. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1388/
[5] Ross CA, Hayden MR. Huntington’s disease. In: Analysis of triplet repeat disorders. Garland Science, 2020; 169-208.
[6] Brown I, Sen A. Neurological disorders. Textbook Occup Med Pract, 2017; 2: 287.[DOI]
[7] Aybek S, Perez DL. Diagnosis and management of functional neurological disorder. BMJ, 2022; 376.[DOI]
[8] Yuhan L, Khaleghi Ghadiri M, Gorji A. Impact of NQO1 dysregulation in CNS disorders. J Transl Med, 2024; 22: 4.[DOI]
[9] Tong H, Yang T, Xu S et al. Huntington’s Disease: Complex Pathogenesis and Therapeutic Strategies. Int J Mol Sci, 2024; 25: 3845.[DOI]
[10] Federico A. Rare neurological diseases: a Pandoraʼs box for neurology (an European and Italian perspective). Rev Neurol-France, 2013; 169: S12-S17.[DOI]
[11] Zhang H, Jiang L, Xian Y et al. Familial hemiplegic migraine type 2: a case report of an adolescent with ATP1A2 mutation. Front Neurol, 2024; 15: 1339642.[DOI]
[12] Pasqui A, Cicaloni V, Tinti L et al. A proteomic approach to investigate the role of the MECP2 gene mutation in Rett syndrome redox regulatory pathways. Arch Biochem Biophys, 2024; 752: 109860.[DOI]
[13] Crowther L, Poms M, Plecko B. Multiomics tools for the diagnosis and treatment of rare neurological disease. J Inherit Metab Dis, 2018; 41: 425-434.[DOI]
[14] Dai X, Shen L. Advances and trends in omics technology development. Front Med-Lausanne, 2022; 9: 911861.[DOI]
[15] Chakraborty D, Sharma N, Kour S et al. Applications of omics technology for livestock selection and improvement. Front Genet, 2022; 13: 774113.[DOI]
[16] Mohammadi-Shemirani P, Sood T, Paré G. From ‘Omics to multi-omics technologies: The discovery of novel causal mediators. Curr Atheroscler Rep, 2023; 25: 55-65.[DOI]
[17] Cano-Gamez E, Trynka G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front Genet, 2020; 11: 424.[DOI]
[18] Chafai N, Bonizzi L, Botti S et al. Emerging applications of machine learning in genomic medicine and healthcare. Crit Rev Cl Lab Sci, 2024: 61: 140-163.[DOI]
[19] Rao M, Gershon MD. The bowel and beyond: the enteric nervous system in neurological disorders. Nat Rev Gastro Hepat, 2016; 13: 517-528.[DOI]
[20] Niesler B, Kuerten S, Demir IE et al. Disorders of the enteric nervous system - a holistic view. Nat Rev Gastro Hepat, 2021; 18: 393-410.[DOI]
[21] Grosu L, Grosu AI, Crisan D et al. Parkinsonʼs disease and cardiovascular involvement: Edifying insights. Biomed Rep, 2023; 18: 1-8.[DOI]
[22] Stewart CB, Ledingham D, Foster VK et al. The longitudinal progression of autonomic dysfunction in Parkinsonʼs disease: A 7-year study. Front Neurol, 2023; 14: 1155669.[DOI]
[23] Tan AH, Chuah KH, Beh YY et al. Gastrointestinal Dysfunction in Parkinson’s Disease: Neuro-Gastroenterology Perspectives on a Multifaceted Problem. J Mov Disord, 2023; 16: 138.[DOI]
[24] Osinalde N, Duarri A, Ramirez J et al. Impaired proteostasis in rare neurological diseases. Semin Cell Dev Biol, 2019; 93: 164-177.[DOI]
[25] Di Resta C, Pipitone GB, Carrera P et al. Current scenario of the genetic testing for rare neurological disorders exploiting next generation sequencing. Neural Regen Res, 2021; 16: 475.[DOI]
[26] Shao N, Zhang H, Wang X et al. Familial hemiplegic migraine type 3 (FHM3) with an SCN1A mutation in a Chinese family: A case report. Front Neurol, 2018; 9: 976.[DOI]
[27] Jain S, Heutink P. From single genes to gene networks: high-throughput-high-content screening for neurological disease. Neuron, 2010; 68: 207-217.[DOI]
[28] Kamboh MI. Genomics and functional genomics of Alzheimerʼs disease. Neurotherapeutics, 2023; 19: 152-172.[DOI]
[29] Ibañez K, Polke J, Hagelstrom RT et al. Whole genome sequencing for the diagnosis of neurological repeat expansion disorders in the UK: a retrospective diagnostic accuracy and prospective clinical validation study. Lancet Neurol, 2022; 21: 234-245.[DOI]
[30] Brittain HK, Scott R, Thomas E. The rise of the genome and personalised medicine. Clin Med, 2017; 17: 545.[DOI]
[31] Rodriguez R, Krishnan Y. The chemistry of next-generation sequencing. Nat Biotechnol, 2023; 41: 1709-1715.[DOI]
[32] Mandlik JS, Patil AS, Singh S. Next-Generation Sequencing (NGS): Platforms and Applications. J Pharm Bioallied Sci, 2024; 16: S41-S45.[DOI]
[33] Heather JM, Chain B. The sequence of sequencers: The history of sequencing DNA. Genomics, 2016; 107: 1-8.[DOI]
[34] Wang F, Xu Y, Wang R et al. TEQUILA-seq: a versatile and low-cost method for targeted long-read RNA sequencing. Nat Commun, 2023; 14: 4760.[DOI]
[35] Arslan S, Garcia FJ, Guo M et al. Sequencing by avidity enables high accuracy with low reagent consumption. Nat Biotechnol, 2024; 42: 132-138.[DOI]
[36] Katara A, Chand S, Chaudhary H et al. Evolution and applications of Next Generation Sequencing and its intricate relations with chromatographic and spectrometric techniques in modern day sciences. J Chromatogr Open, 2024; 5: 100121.[DOI]
[37] Kwon R, Yeung CCS. Advances in next-generation sequencing and emerging technologies for hematologic malignancies. Haematologica, 2024; 109: 379-387.[DOI]
[38] Maheshwari K, Musyuni P, Moulick A et al. Unveiling the microbial symphony: Next-Gen sequencing and bioinformatics insights into the human gut microbiome. Health Sci Rev, 2024; 11: 100173.[DOI]
[39] Pevsner J. Introduction: BLAST: Basic Local Alignment Search Tool. In: Bioinformatics and Functional Genomics. John Wiley & Sons Publishing: USA, 2009; 100-138.
[40] Meza-Sosa KF, Valle-Garcia D, González-Conchillos H et al. Molecular Mimicry between Toxoplasma gondii B-Cell Epitopes and Neurodevelopmental Proteins: An Immunoinformatic Approach. Biomolecules, 2024; 14: 933.[DOI]
[41] James LM, Strickland Z, Lopez N et al. Identification and Analysis of Axolotl Homologs for Proteins Implicated in Human Neurodegenerative Proteinopathies. Genes, 2024; 15: 310.[DOI]
[42] Jia N, Yu S, Zhang G et al. Recurrent MECR R258W causes adult-onset optic atrophy: A case report. Eur J Med Genet, 2024; 68: 104917.[DOI]
[43] Ghasemi MR, Sadeghi H, Hashemi-Gorji F et al. Exome sequencing reveals neurodevelopmental genes in simplex consanguineous Iranian families with syndromic autism. BMC Med Genomics, 2024; 17: 196.[DOI]
[44] Heng Li, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009; 25: 1754-1760.[DOI]
[45] Horváth V, Garza R, Jönsson ME et al. Mini-heterochromatin domains constrain the cis-regulatory impact of SVA transposons in human brain development and disease. Nat Struct Mol Biol, 2024 [Epub ahead of print].
[46] Zhang T, Xue Y, Su S et al. RNA-binding protein Nocte regulates Drosophila development by promoting translation reinitiation on mRNAs with long upstream open reading frames. Nucleic Acids Res, 2023; 52: 885-905.[DOI]
[47] Langmead B, Trapnell C, Pop M et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol, 2009; 10: R25.[DOI]
[48] McKenna A, Hanna M, Banks E et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 2010; 20: 1297-1303. DOI: 10.1101/gr.107524.110
[49] Danecek P BJ, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H. . Twelve years of SAMtools and BCFtools. 2021. DOI: 10.1093/gigascience/giab008.
[50] Skoczylas S, Jakiel P, Płoszaj T et al. Novel potentially pathogenic variants detected in genes causing intellectual disability and epilepsy in Polish families. Neurogenetics, 2023; 24: 221-229.[DOI]
[51] Hu T, Chitnis N, Monos D et al. Next-generation sequencing technologies: An overview. Hum Immunol, 2021; 82: 801-811.[DOI]
[52] Satam H, Joshi K, Mangrolia U et al. Next-generation sequencing technology: Current trends and advancements. Biology, 2023; 12: 997.[DOI]
[53] Austin-Tse CA, Jobanputra V, Perry DL et al. Best practices for the interpretation and reporting of clinical whole genome sequencing. Npj Genom Med, 2022; 7: 27.[DOI]
[54] Ross JP, Dion PA, Rouleau GA. Exome sequencing in genetic disease: recent advances and considerations. F1000 Res, 2020; 9: 336.[DOI]
[55] Pei XM, Yeung MHY, Wong ANN et al. Targeted sequencing approach and its clinical applications for the molecular diagnosis of human diseases. Cells, 2023; 12: 493.[DOI]
[56] Vestergaard LK, Oliveira DN, Høgdall CK et al. Next generation sequencing technology in the clinic and its challenges. Cancers, 2021; 13: 1751.[DOI]
[57] Logsdon GA, Vollger MR, Eichler EE. Long-read human genome sequencing and its applications. Nat Rev Genet, 2020; 21: 597-614.[DOI]
[58] van der Reis AL, Beckley LE, Olivar MP et al. Nanopore short‐read sequencing: A quick, cost‐effective and accurate method for DNA metabarcoding. Environ DNA, 2023; 5: 282-296.[DOI]
[59] Amarasinghe SL, Su S, Dong X et al. Opportunities and challenges in long-read sequencing data analysis. Genome Biol, 2020; 21: 1-16.[DOI]
[60] Ben Khedher M, Ghedira K, Rolain JM et al. Application and challenge of 3rd generation sequencing for clinical bacterial studies. Int J Mol Sci, 2022; 23: 1395.[DOI]
[61] Turro E, Astle WJ, Megy K et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature, 2020; 583: 96-102.[DOI]
[62] Oh JH, Jo S, Park KW et al. Whole-genome sequencing reveals an association between small genomic deletions and an increased risk of developing Parkinson’s disease. Exp Mol Med, 2023; 55: 555-564.[DOI]
[63] Chi CS, Tsai CR, Lee HF. Resolving unsolved whole-genome sequencing data in paediatric neurological disorders: a cohort study. Arch Dis Child, 2024; 109: 730-735.[DOI]
[64] Bagger FO, Borgwardt L, Jespersen AS et al. Whole genome sequencing in clinical practice. BMC Med Genomics, 2024; 17: 39.[DOI]
[65] Funayama M, Nishioka K, Li Y et al. Molecular genetics of Parkinson’s disease: Contributions and global trends. J Hum Genet, 2023; 68: 125-130.[DOI]
[66] Tolosa E, Garrido A, Scholz SW et al. Challenges in the diagnosis of Parkinson's disease. Lancet Neurol, 2021; 20: 385-397.[DOI]
[67] Aaltio J, Hyttinen V, Kortelainen M et al. Cost-effectiveness of whole-exome sequencing in progressive neurological disorders of children. Eur J Paediatr Neuro, 2022; 36: 30-36.[DOI]
[68] Jo YH, Choi SH, Yoo HW et al. Clinical use of whole exome sequencing in children with developmental delay/intellectual disability. Pediatr Neonatol, 2024; 65: 445-450.[DOI]
[69] Chandy T. Intervention of next-generation sequencing in diagnosis of Alzheimer’s disease: challenges and future prospects. Dement Neuropsych, 2023; 17: e20220025.[DOI]
[70] Glotov OS, Chernov AN, Glotov AS. Human Exome Sequencing and Prospects for Predictive Medicine: Analysis of International Data and Own Experience. J Pers Med, 2023; 13: 1236.[DOI]
[71] Li Q, Chen W, Wang C et al. Whole-exome sequencing reveals common and rare variants in immunologic and neurological genes implicated in achalasia. Am J Hum Genet, 2021; 108: 1478-1487.[DOI]
[72] Alvarez-Mora MI, Rodríguez-Revenga L, Jodar M et al. Implementation of Exome Sequencing in Clinical Practice for Neurological Disorders. Genes, 2023; 14: 813.[DOI]
[73] Sheth F, Shah J, Jain D et al. Comparative yield of molecular diagnostic algorithms for autism spectrum disorder diagnosis in India: Evidence supporting whole exome sequencing as first tier test. BMC Neurol, 2023; 23: 292.[DOI]
[74] Cheroni C, Caporale N, Testa G. Autism spectrum disorder at the crossroad between genes and environment: contributions, convergences, and interactions in ASD developmental pathophysiology. Mol Autism, 2020; 11: 69.[DOI]
[75] Ichikawa S, Yergert K, Gasser B et al. O34: Utility of targeted RNA analysis in neurological disorders. Genet Med Open, 2024; 2: 101473.[DOI]
[76] Hac NEF, Gold DR. Advances in diagnosis and treatment of vestibular migraine and the vestibular disorders it mimics. Neurotherapeutics, 2024; 21: e00381.[DOI]
[77] Bacquet J, Stojkovic T, Boyer A et al. Molecular diagnosis of inherited peripheral neuropathies by targeted next-generation sequencing: molecular spectrum delineation. BMJ Open, 2018; 10.1136/bmjopen-2018-021632
[78] Schalock RL, Luckasson R, Tassé MJ. An overview of intellectual disability: Definition, diagnosis, classification, and systems of supports. Ajidd-Am J Intellect, 2021; 126: 439-442.[DOI]
[79] Ilyas M, Mir A, Efthymiou S et al. The genetics of intellectual disability: advancing technology and gene editing. F1000 Res, 2020; 9: 22.[DOI]
[80] Lee K, Cascella M, Marwaha R. Intellectual Disability. StatPearls Publishing: Treasure Island, USA, 2023.
[81] Jansen S, Vissers LE, de Vries BB. The Genetics of Intellectual Disability. Brain Sci, 2023; 13: 231.[DOI]
[82] Higuchi Y, Takashima H. Clinical genetics of Charcot-Marie-Tooth disease. J Hum Genet, 2023; 68: 199-214.[DOI]
[83] Andrews SJ, Renton AE, Fulton-Howard B et al. The complex genetic architecture of Alzheimer's disease: novel insights and future directions. EBiomedicine, 2023; 90: 104511.[DOI]
[84] Kim JJ, Vitale D, Otani DV et al. Multi-ancestry genome-wide association meta-analysis of Parkinson’s disease. Nat Genet, 2024; 56: 27-36.[DOI]
[85] Lin F, Li J, Wang Z et al. Replication of previous autism-GWAS hits suggests the association between NAA1, SORCS3, and GSDME and autism in the Han Chinese population. Heliyon, 2024; 10: e23677.[DOI]
[86] Uffelmann E, Huang QQ, Munung NS et al. Genome-wide association studies. Nat Rev Method Prime, 2021; 1: 59.[DOI]
[87] Tan MM, Lawton MA, Jabbari E et al. Genome‐wide association studies of cognitive and motor progression in Parkinson's disease. Movement Disord, 2021; 36: 424-433.[DOI]
[88] Mullins N, Forstner AJ, O’Connell KS et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet, 2021; 53: 817-829.[DOI]
[89] Grenn FP, Kim JJ, Makarious MB et al. The Parkinson's disease genome‐wide association study locus browser. Movement Disord, 2020; 35: 2056-2067.[DOI]
[90] Escott-Price V, Hardy J. Genome-wide association studies for Alzheimer’s disease: bigger is not always better. Brain Commun, 2022; 4: fcac125.[DOI]
[91] Koretsky MJ, Alvarado C, Makarious MB et al. Genetic risk factor clustering within and across neurodegenerative diseases. Brain, 2023; 146: 4486-4494.[DOI]
[92] Wang W, Gao R, Yan X et al. Relationship between plasma brain-derived neurotrophic factor levels and neurological disorders: An investigation using Mendelian randomisation. Heliyon, 2024; 10: e30415.[DOI]
[93] Babu M, Snyder M. Multi-Omics Profiling for Health. Mol Cell Proteomics, 2023; 22: 100561.[DOI]
[94] Chen C, Wang J, Pan D et al. Applications of multi-omics analysis in human diseases. MedComm, 2023; 4: e315.[DOI]
[95] O'Connor LM, O'Connor BA, Lim SB et al. Integrative multi-omics and systems bioinformatics in translational neuroscience: A data mining perspective. J Pharm Anal, 2023; 13: 836-850.[DOI]
[96] Zhao N, Quicksall Z, Asmann YW et al. Network approaches for omics studies of neurodegenerative diseases. Front Genet, 2022; 13: 984338.[DOI]
[97] Le Grand Q, Tsuchida A, Koch A et al. Diffusion imaging genomics provides novel insight into early mechanisms of cerebral small vessel disease. Mol Psychiatr 2024 [Online ahead of print]. https://doi.org/10.1038/s41380-024-02604-7
[98] Shaath R, Al-Maraghi A, Ali H et al. Integrating Genome Sequencing and Untargeted Metabolomics in Monozygotic Twins with a Rare Complex Neurological Disorder. Metabolites, 2024; 14: 152.[DOI]
[99] van Karnebeek CDM, Tarailo-Graovac M, Leen R et al. CIAO1 and MMS19 deficiency: A lethal neurodegenerative phenotype caused by cytosolic Fe-S cluster protein assembly disorders. Genet Med, 2024; 26: 101104.[DOI]
[100] Jiang D, Nan H, Chen Z et al. Genetic insights into drug targets for sporadic Creutzfeldt-Jakob disease: Integrative multi-omics analysis. Neurobiol Dis, 2024; 199: 106599.[DOI]
[101] Pascual-Alonso A, Xiol C, Smirnov D et al. Multi-omics in MECP2 duplication syndrome patients and carriers. Eur J Neurosci, 2024; 60: 4004-4018.[DOI]
[102] Segarra-Casas A, Yépez VA, Demidov G et al. An Integrated Transcriptomics and Genomics Approach Detects an X/Autosome Translocation in a Female with Duchenne Muscular Dystrophy. Int J Mol Sci, 2024; 25: 7793.[DOI]
[103] Jonson C, Levine KS, Lake J et al. Assessing the lack of diversity in genetics research across neurodegenerative diseases: A systematic review of the GWAS Catalog and literature. Alzheimer's & Dementia, 2024; 20: 5740-5756. DOI: 10.1002/alz.13873
[104] Rutten-Jacobs L, McIver T, Reyes A et al. Racial and ethnic diversity in global neuroscience clinical trials. Cont Clin Trial Comm, 2024; 37: 101255.[DOI]
[105] Khani M, Cerquera-Cleves C, Kekenadze M et al. Towards a Global View of Parkinson's Disease Genetics. Ann Neurol, 2024; 95: 831-842.[DOI]
[106] Tanaka M, Vécsei L. A Decade of Dedication: Pioneering Perspectives on Neurological Diseases and Mental Illnesses. Biomedicines, 2024; 12: 1083.[DOI]
[107] Mavroudis I, Kazis D, Kamal FZ et al. Understanding Functional Neurological Disorder: Recent Insights and Diagnostic Challenges. Int J Mol Sci, 2024; 25: 4470.[DOI]
[108] Caznok Silveira AC, Antunes ASLM, Athié MCP et al. Between neurons and networks: investigating mesoscale brain connectivity in neurological and psychiatric disorders. Front Neurosci, 2024; 18: 1340345.[DOI]
[109] Mayston MJ, Saloojee GM, Foley SE. The Bobath Clinical Reasoning Framework: A systems science approach to the complexity of neurodevelopmental conditions, including cerebral palsy. Dev Med Child Neurol, 2024; 66: 564-572.[DOI]
[110] Tripathi A, Pandey VK, Sharma G et al. Genomic Insights into Dementia: Precision Medicine and the Impact of Gene-Environment Interaction. Aging Dis, 2024; 15: 2113-2135.[DOI]
[111] Koloi-Keaikitse S, Kasule M, Kwape I et al. Understanding cultural values, norms and beliefs that may impact participation in genome-editing related research: Perspectives of local communities in Botswana. Dev World Bioeth, 2024.[DOI]
[112] van der Geest MA, Maeckelberghe ELM, van Gijn ME et al. Systematic reanalysis of genomic data by diagnostic laboratories: a scoping review of ethical, economic, legal and (psycho)social implications. Eur J Hum Genet, 2024; 32: 489-497.[DOI]
[113] Olorunsogo TO, Balogun OD, Ayo-Farai O et al. Bioinformatics and personalized medicine in the U.S.: A comprehensive review: Scrutinizing the advancements in genomics and their potential to revolutionize healthcare delivery. World J Adv Res Rev, 2024; 21: 335-351.[DOI]
[114] Sahan K, Lyle K. Response to commentaries: ethical preparedness in genomic medicine how NHS clinical scientists navigate ethical issues. J Med Ethics, 2024; 50: 532.[DOI]
[115] Nevin SM, Le Marne FA, Beavis E et al. Psychosocial experiences of clinicians providing care for children with severe neurological impairment. Dev Med Child Neurol, 2024; 66: e209-e209.[DOI]
[116] Yi H, Trivedi MS, Crew KD et al. Understanding Social, Cultural, and Religious Factors Influencing Medical Decision-Making on BRCA1/2 Genetic Testing in the Orthodox Jewish Community. Public Health Genom, 2024; 27: 57-67.[DOI]
[117] Gaydarska H, Takashima K, Shahrier S et al. The interplay of ethics and genetic technologies in balancing the social valuation of the human genome in UNESCO declarations. Eur J Hum Genet, 2024; 32: 725-730.[DOI]
[118] Nankya H, Wamala E, Alibu VP et al. Community engagement in genetics and genomics research: a qualitative study of the perspectives of genetics and genomics researchers in Uganda. BMC Med Ethics, 2024; 25: 1.[DOI]
[119] Boon P, Lescrauwaet E, Aleksovska K et al. A strategic neurological research agenda for Europe: Towards clinically relevant and patient-centred neurological research priorities. Eur J Neurol, 2024; 31: e16171.[DOI]
[120] Horn R, Merchant J, Horn R et al. Ethical and social implications of public–private partnerships in the context of genomic/big health data collection. Eur J Hum Genet, 2024; 32: 736-741.[DOI]
[121] Strianese O, Rizzo F, Ciccarelli M et al. Precision and personalized medicine: how genomic approach improves the management of cardiovascular and neurodegenerative disease. Genes, 2020; 11: 747.[DOI]
[122] Sanchez-Luquez KY, Carpena MX, Karam SM et al. The contribution of whole-exome sequencing to intellectual disability diagnosis and knowledge of underlying molecular mechanisms: A systematic review and meta-analysis. Mutat Res Rev Mutat Res, 2022; 790: 108428.[DOI]
[123] Mitsuhashi S, Matsumoto N. Long-read sequencing for rare human genetic diseases. J Hum Genet, 2020; 65: 11-19.[DOI]
[124] Tirrell KM, O'Neill HC. Comparing the diagnostic and clinical utility of WGS and WES with standard genetic testing (SGT) in children with suspected genetic diseases: A systematic review and meta-analysis. Medrxiv 2023 [Epub ahead of print]. https://doi.org/10.1101/2023.07.17.23292722
[125] Ormond KE, Wheeler MT, Hudgins L et al. Challenges in the clinical application of whole-genome sequencing. Lancet, 2010; 375: 1749-1751.[DOI]
[126] Xuan J, Yu Y, Qing T et al. Next-generation sequencing in the clinic: promises and challenges. Cancer Lett, 2013; 340: 284-295.[DOI]
[127] Dong X, Liu C, Dozmorov M. Review of multi-omics data resources and integrative analysis for human brain disorders. Brief Funct Genomics, 2021; 20: 223-234.[DOI]
[128] Clark C, Dayon L, Masoodi M et al. An integrative multi-omics approach reveals new central nervous system pathway alterations in Alzheimer’s disease. Alzh Res Ther, 2021; 13: 1-19.[DOI]
[129] Nativio R, Lan Y, Donahue G et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat Genet, 2020; 52: 1024-1035.[DOI]
[130] Alvarado CX, Makarious MB, Weller CA et al. omicSynth: An open multi-omic community resource for identifying druggable targets across neurodegenerative diseases. Am J Hum Genet, 2024; 111: 150-164.[DOI]
Brief of Corresponding Author(s)
Roy D. Sleator He is the Professor of Biological Sciences at Munster Technological University, Ireland. He holds a BSc in Microbiology, an MA in Teaching & Learning in Higher Education, and a PhD in Molecular Biology from University College Cork, Ireland. He also holds a PGC in Bioinformatics from the University of Manchester, UK, as well as an MBA from Anglia Ruskin University, UK, and a DSc (Higher Doctorate) on published works from the National University of Ireland. Sleator has published over 200 peer reviewed papers. He has more than 11,000 citations and an h-index of 53. |



