Covalent Flexible Peptide Docking in Drug Discovery: Current Challenges and Potential Interventions
Zineb Lafifi1, Imane Bjij1,2, Mahmoud E.S. Soliman1*
1Molecular Bio-computation and Drug Design Laboratory, School of Health Sciences, University of KwaZulu-Natal, Westville Campus, Durban, South Africa
2Institut Supérieur des Professions Infirmières et Techniques de Santé, Dakhla, Morocco
*Correspondence to: Mahmoud E.S. Soliman, PhD, Professor, Molecular Bio-computation and Drug Design Laboratory, School of Health Sciences, University of KwaZulu-Natal, Westville Campus, Durban, 4000, South Africa; Email: soliman@ukzn.ac.za
DOI: 10.53964/id.2024015
Abstract
Recently, there has been a significant increase in the search for new covalent inhibitors through drug discovery platforms, which has led to the development and implementation of new computational tools, including covalent docking methods to predict the binding mode and affinity of covalent ligands. Since the discovery of insulin nearly a century ago, more than 80 peptide medications have hit the market to treat a wide range of diseases, including diabetes, cancer, osteoporosis, multiple sclerosis, HIV infection, and chronic pain. Electrophilic peptides that form covalent bonds with target proteins have great potential for binding targets that have been previously considered undruggable. Despite the recent advancements in computational performance and docking algorithms, covalent docking still poses a number of challenges. Peptides covalent docking presents additional challenges, the main ones being the choice of the optimal peptide sequence, incorporation of electrophilic warheads, the inherent flexibility of peptide structures, and the fact that, unlike small molecules, peptides do fold. In this review, we present a brief overview of the current state of peptide therapeutics in drug discovery, covalent docking in general, covalent peptide binders, and-with particular emphasis-the difficulties that covalent peptide-protein docking is currently facing and some potential solutions.
Keywords: covalent docking, flexible docking, peptide covalent docking, covalent inhibition, peptides, irreversible inhibition
1 INTRODUCTION
Even though covalent inhibitors have been used therapeutically for more than a century, there was widespread opposition in the pharmaceutical industry to their further development due to safety worries. After the creation of a wide range of covalent inhibitors for the treatment of human health conditions and the FDA’s approval of several covalent therapeutics for use in humans, this disposition has recently been reversed[1]. In the past few years, there has been a significant increase in the search for new covalent inhibitors[1].
Recent developments have also made it possible to use them as chemical probes to reveal new and harder-to-reach targets. The rising interest in covalent drug discovery prompted the development of new computational tools, such as covalent docking techniques, that are available to predict the binding mode and affinity of covalent ligands[1-3]. As a result of this expanding research into the discovery of covalent inhibitors, more than 80 covalent therapeutics have been approved by the FDA and currently about 30% of the marketed drugs are covalent inhibitors[1].
Figure 1 displays some selected examples of covalent drugs that the FDA has approved and the year of approval. Acetylsalicylic acid, also known as aspirin, was the first covalent inhibitor to be commercialized in 1899, and it covalently binds to Ser-530 of prostaglandin endoperoxide synthase-1 via its acetyl warhead[2]. This was followed by the development of a vast array of covalent drugs for various disease. For instance, timoprazole demonstrated suppression of stomach acid secretion in 1975. However, further development of timoprazole was halted due to its toxicity[3]. Omeprazole, a derivative of timoprazole, was then identified in 1979 as the first medication class currently referred to as a proton pump inhibitor (PPI)[4]. Omeprazole covalently binds to hydrogen-potassium ATPase through disulphide bond. Lansoprazole, which was then introduced to the market in the early 1990s, and given FDA approval in 1995, hence was the second PPI medication to enter the market[2]. Two of the most popular covalent medications in the 1990s were clopidogrel and fosfomycin. Clopidogrel is an antiplatelet drug that attaches to the P2Y21 purinergic receptor irreversibly through the -SH group[5]. Through the inhibition of the enzyme MurA, fosfomycin suppresses the synthesis of bacterial cell wall. It is equipped with an epoxide warhead aimed against MurA’s active site residue Cys115[6]. One of the rare covalent inhibitors with a boronic acid warhead is the anticancer drug brentezomib. In 2003, it received approval and released to the market. Two of the most recently approved covalent inhibitors of BTK and EGFR are Zanubrutinib (approved November 2019, marketed under the trade name Brukinsa®)[7] and dacomitinib (approved September 2018, marketed under the name Vizimpro®)[8].
|
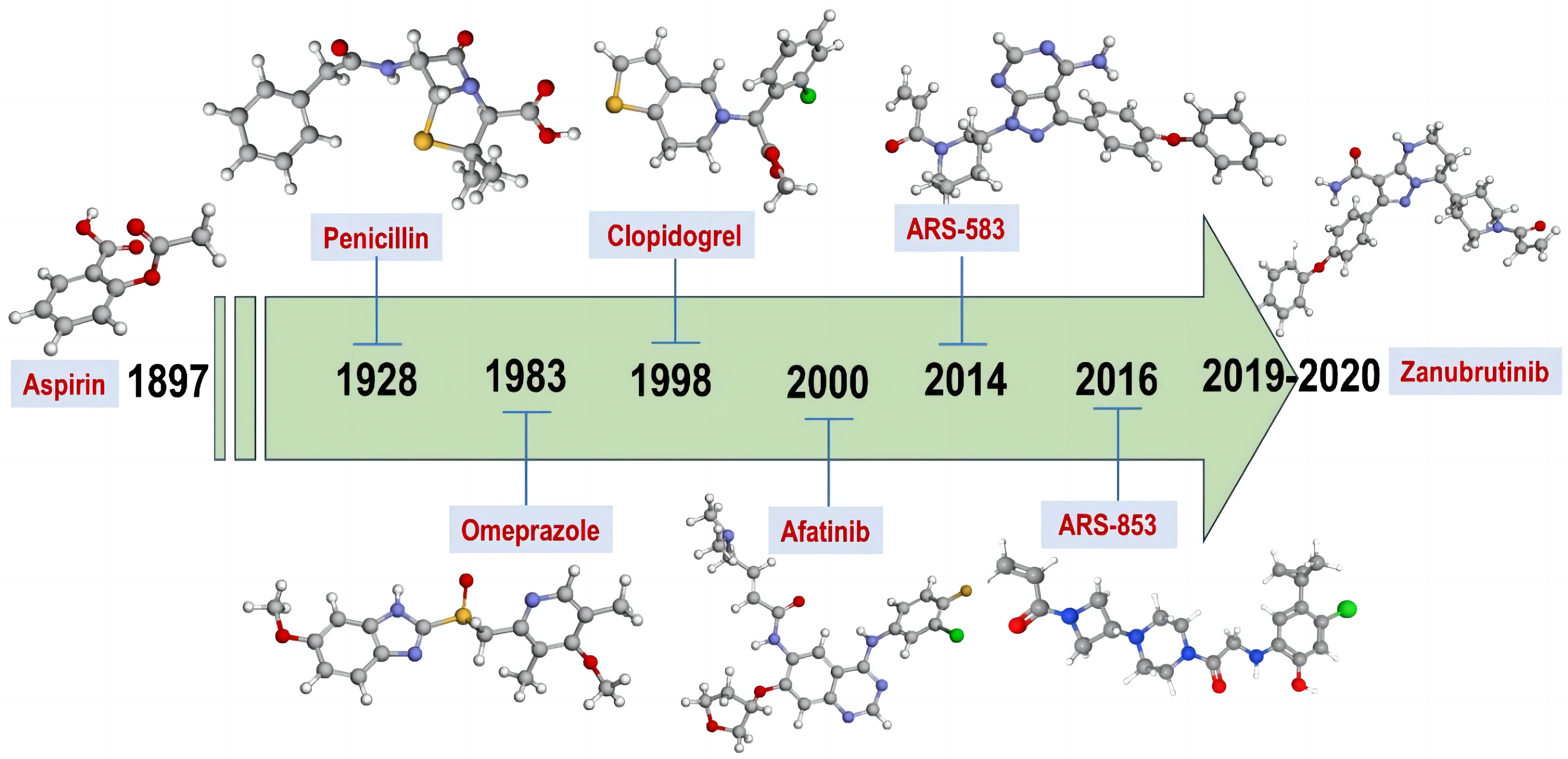
Figure 1. Some Examples of the FDA-Approved Covalent Drugs and the Year in Which It Was Approved.
1.1 Small Molecules Inhibitors vs. Peptides – Evolution of Covalent Peptides
Small molecules are typically used to target particular proteins, but in many instances, due to their size, flatness, and lack of natural substrates, some targets are notoriously challenging to drug using small molecules. Sites of protein-protein interactions, shallow allosteric pockets, transcription factors, and DNA binding proteins generally are examples of difficult protein targets[2]. Using peptides, which have a much larger surface area and can interact with “hot-spots” on the target’s binding surface, is a common strategy for attacking such targets. Such peptides have a high degree of biological affinity and target specificity and safety[9-11]. 1922 was the starting year of the field of peptide drugs, using animal insulin extracted from bovine and porcine pancreas to treat the type 1 diabetes[12]. Then, in 1954, the team led by Vincent Du Vigneaud introduced oxytocin as the first artificial polypeptide[13]. Bruce Merrifield took the next step, believing that peptide synthesis could be automated by using a solid phase to assemble different amino acids. This research led to the creation of Solid Phase Peptide Synthesis in 1963[14].
Although not usually in accordance with Lipinski’s rule of five, recent chemical methodologies have been shown to enhance the pharmacokinetic properties of peptides, such as bioavailability, permeability, and in vivo stability. These chemical modifications may include N-methylation, cyclization, and incorporation of d-amino acids[15-17]. It is still difficult to find high-affinity peptide binders that can compete with natural cellular interactions and get around the inherent binding challenges of large protein surfaces[11] Longer chain peptides might be able to interact with more protein “hot spots”, but doing so will increase the binding enthalpy and raise the entropic cost, which will have a significant detrimental effect on the binding affinity. Additionally, pharmacokinetic profiles for longer chain peptides are usually poor. Forming a covalent bonding between the peptide and its target is one possible approach that might be used to overcome this challenge, hence greatly increase the peptide’s potency[18,19] However, compared to covalent small molecule inhibitors, the reported number of covalent peptides is still much lower. Although significant efforts have been made to aid the modelling and design of non-covalent peptides, the molecular simulations of covalent peptide interactions remain a challenging task, especially with the lack of peptide-specific validated protocol that takes into account covalent binding between the peptide and the receptor[18,20-22]. Along with our earlier reports on covalent docking[23-29], The current report will concentrate on covalent peptide docking, its challenges, and provide potential solutions including technical guidance to useful tools and approaches to assist with the rational design of covalent peptide inhibitors.
2 COVALENT VS. NON-COVALENT INHIBITION
In general, covalent inhibitors bind to the targeted protein and inactivate the protein temporarily or permanently[30]. In fact, the mechanism of the inhibition process depends on the reaction equilibrium between the ligand functional groups - that can be electrophilic or nucleophilic and the amino acids of the targeted protein[31]. As a result of this nature of interactions between the “warhead” and the protein active site, the complex can represent a bound or unbound states[32], which results to one of two scenarios: covalent or non-covalent interactions[33] (Figure 2).
|
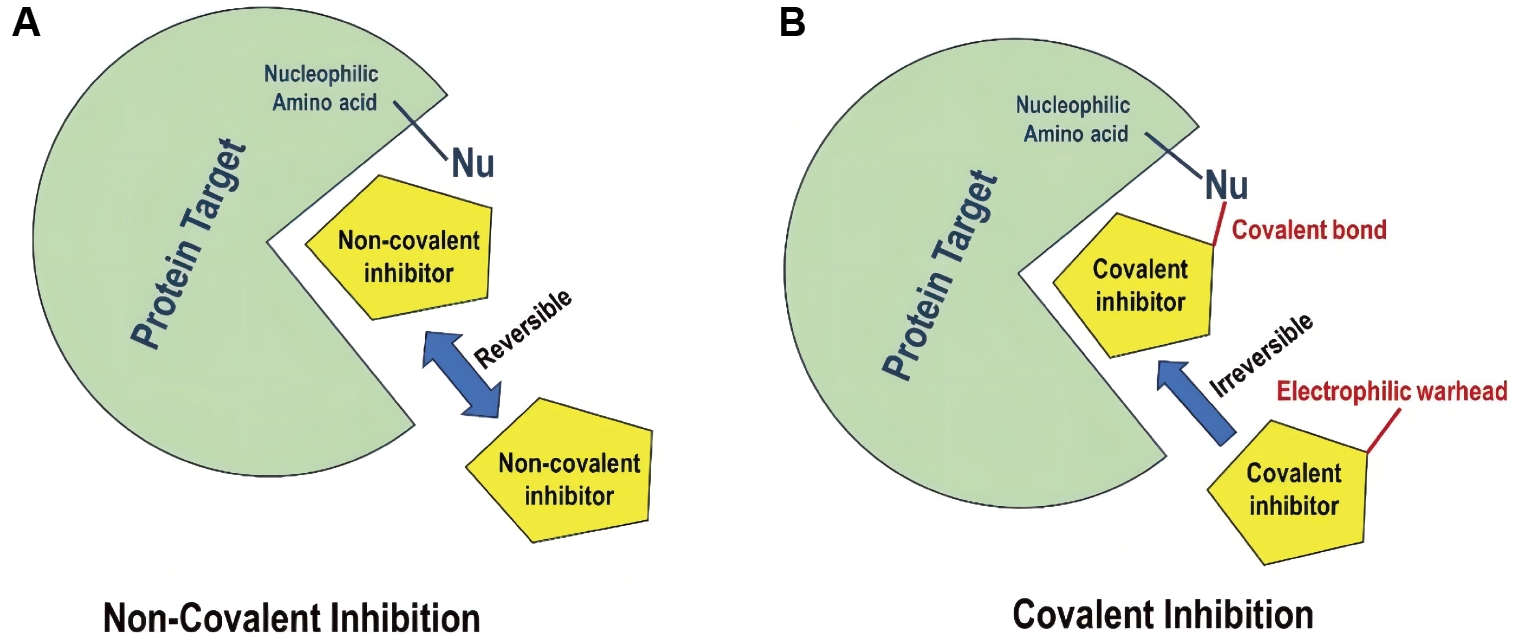
Figure 2. Graphical Illustration of Non-Covalent Inhibition (A) vs. Covalent Binding (B).
Interestingly, it was demonstrated that different drugs can bind with the same target via different mechanisms. For instance, pirtobrutinib, the first and the only non-covalent BTK inhibitor approved by FDA recently[34,35], was found to bind non-covalently to BTK (Figure 3A), while zanubrutinib represents an example of covalent binder to the same protein, BTK (Figure 3B)[36].
|
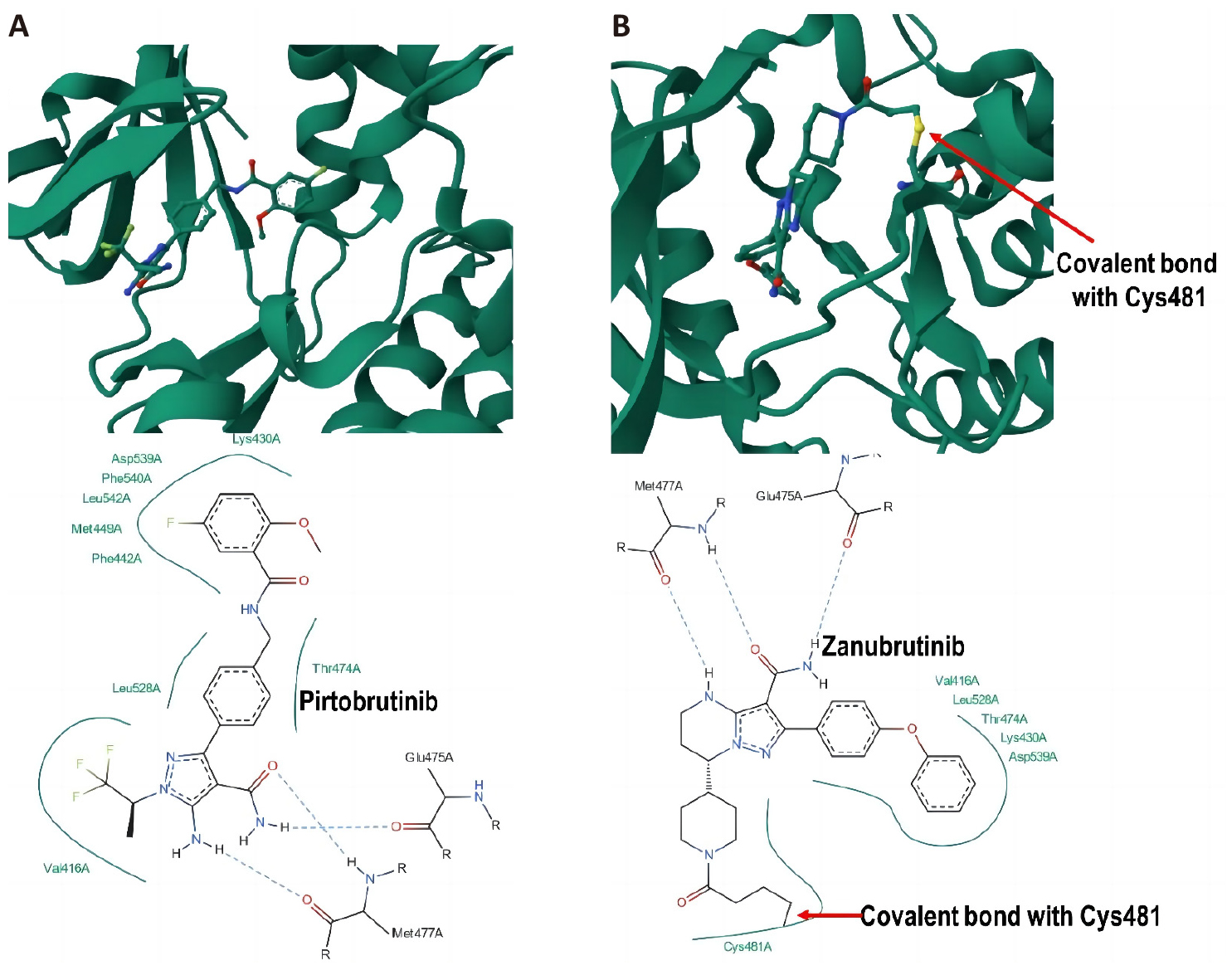
Figure 3. 3D Illustration and Corresponding 2D Interaction Plot of (A) Non-Covalent Interactions of Pirtobrutinib With BTK (PDB Code: 8FLL) and (B) Covalent Interaction of Zanubrutinib With BTK (PDB Code: 6J6M).
2.1 Covalent Inhibition Was Always Thought to Be “Irreversible” – Is This Still the Case?
Generally speaking, covalent inhibitors are compounds that, by design, are meant to form a covalent bond with a particular molecular target. It was always thought that covalent inhibitors are irreversible until the concept of covalent-reversible modification was recently introduced to the for several biological targets[37-42]. In order to maintain the desired maximum drug-target residence time on the target of interest, it is hoped that this modification to reversible nature will lessen the risk of unspecific covalent modification and, consequently, possibly lower the risk of severe side effects.
Depending on the selected warhead, the covalent bond may be either reversible or irreversible. Rauh et al have developed an efficient methodology to characterize the covalent and reversible characteristics of a library of covalent reversible inhibitors and investigated their potential in targeting tyrosine kinase EGFR[43]. The electron-withdrawing substituents in these compounds make the β-keto position more liable to nucleophilic attack, hence quickening the adding process in order to bind the target protein covalently. Furthermore, these substituents make the Cα-H in the covalently attached inhibitor more acidic, which makes it easier to quickly remove the proton when the protein-inhibitor environment changes[44,45]. Figure 4 illustrates an example of reversible covalent inhibitor HKI-272 in a complex with EGFR kinase domain T790M/L858R mutant[46].
|
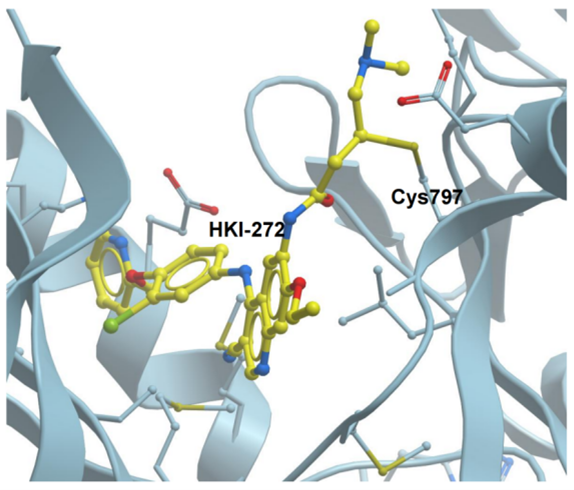
Figure 4. Crystal Structures of HKI-272 in a Complex With EGFR Kinase Domain T790M/L858R Mutant (PDB: 3W2Q) Via Covalent Irreversible Interactions.
3 COVALENT DOCKING OF PEPTIDES IN DRUG DISCOVERY – NOT TRIVIAL!
We have previously discussed covalent docking in detail, including its uses and applications, available tools, advantages and disadvantages[25-27]. To avoid repetition of information, the primary objective of this review is the covalent docking technique specifically related to peptide-based molecules. But first, a number of important aspects must be thoroughly grasped before discussing covalent docking approach for peptides. These include the choice of amino acid sequence, the structural and chemical prerequisites for a peptide to create a covalent link with a target protein, flexibility and so on. In the following text, we discuss each of these aspects, which all together would help build up a sense of understanding of rational design of peptide-based covalent inhibitors.
3.1 Peptide Structural Requirements – Peptide Sequence, Electrophilic Warhead Flexibility and Folding Challenges
3.1.1 Rational Design of Peptide Sequence
The physicochemical and ADMET qualities of a peptide are determined by the amino acid sequence that forms it. The physicochemical characteristics of peptide-based medications can be tailored using a variety of techniques to overcome issues such as low permeability, metabolic instability, and limited tissue residence duration[47]. In this context, the use of bioinformatics, artificial intelligence, and computational modeling applications has been crucial48]. The variety and order of amino acids can be intelligently chosen based on the research goal. Here we present some useful tools that help with the prediction of peptide sequence, physicochemical and toxicity profiles of the designed peptides. Peptide combination generator server (https://pepcogen.bicfri.in/) is one useful tool that can be used to create any combination that a specific peptide sequence based on its physiochemical characteristics. ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/index.html) is an in silico method, which is developed to predict and design toxic/non-toxic peptides. Database of Antimicrobial Activity and Structure of Peptides (https://dbaasp.org/) is the database that is manually curated. It is created to give scientists the knowledge and analytical tools they need to specifically create antimicrobial compounds with a high therapeutic index. PEP-FOLD (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD/) is a de novo approach aimed at predicting peptide structures from amino acid sequences. This approach combines the projected series of SA letters to a greedy algorithm and a coarse-grained force field, based on the structural alphabet SA letters to characterize the conformations of four successive residues. PrMFTP (http://bioinfo.ahu.edu.cn/PrMFTP/) is another a web application used to identify multi-functionalities of therapeutic Peptides. More precisely, PrMFTP learns and fully extracts informative features from peptide sequences in order to anticipate multifunctional therapeutic peptides by utilizing multi-scale convolutional neural networks, bi-directional long short-term memory, and multi-head self-attention techniques.
3.1.2 Electrophilic Reactive Warhead
A warhead refers to the chemical moiety that forms a covalent bond with the target protein through the classical electrophilic-neutrophilic reaction with nucleophilic amino acids in the protein binding site such as cysteine, lysine etc. Selecting the right warhead or modification is essential since it maximizes the inhibitor’s effectiveness, reactivity, and selectivity with the target protein while reducing its off-target effects[49-51]. In contrast to small molecules, which might naturally have electrophilic groups in their chemical structures, peptides lack an electrophilic moiety by nature, hence it is necessary to add one in order for a peptide to form a covalent connection with a nucleophilic amino acid, most commonly cysteine and lysine, in the binding site of target protein. Common examples of warheads include acrylamides, thioesters, alkynylbenzaldehydes, sulfanamides, and boronate-based reagents, among others are shown in Figure 5.
|
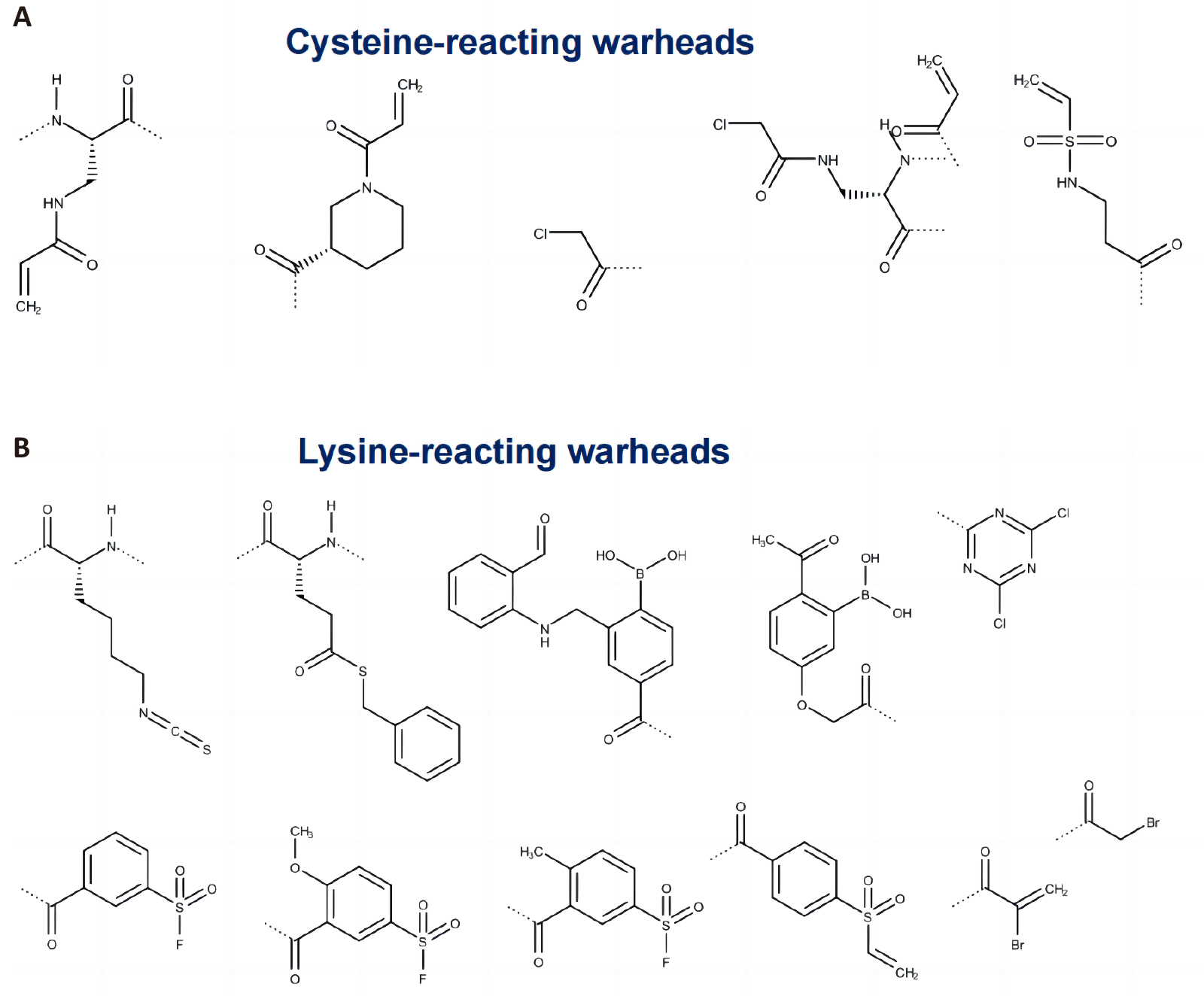
Figure 5. Common Examples of Warhead Chemical Structures That Are Used as Peptide Modifiers: (A) Cysteine-Reacting and (B) Lysine-Reacting. The dotted line in the structures above is the attachment point to peptide chain.
Determining the precise chemical groups that will interact with the target protein is necessary for choosing the reactive electrophilic moiety in peptide design[52]. Example of chemical modification of peptides to include electrophilic warhead is shown in Figure 6.
|
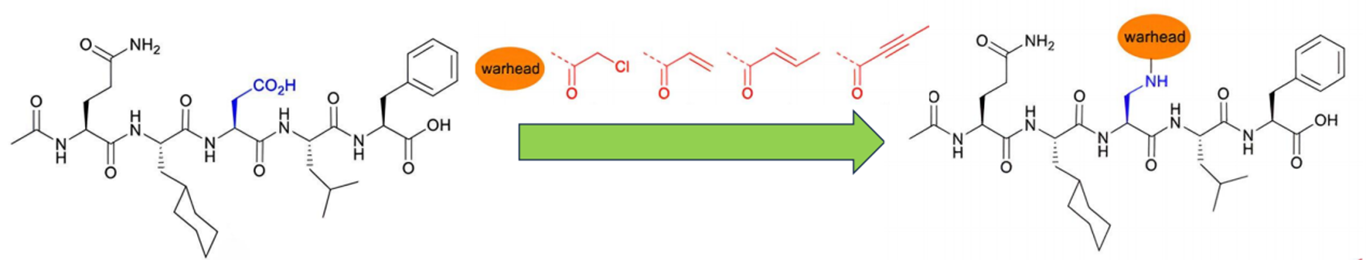
Figure 6. An Example of Chemical Modifications of Peptides to Incorporate an Electrophilic Warhead. The 2D chemical structures were taken from[53].
In this regard, CovPepDock (https://rosie.graylab.jhu.edu/cov_pep_dock) is a useful predictive tool that can be used to identify an optimal warhead and create peptide covalent modifiers (warheads) by using a known non-covalent binder as a starting point to make irreversible covalent link with a target cysteine[54].
3.1.3 Flexibility Challenge
Unlike small molecules, besides their conformational flexibility, peptides often have conformational preferences. Many short peptides in solution can adapt their native secondary structures, α helices, and β hairpins[55]. The fact that peptides have inherent conformational flexibility must therefore be taken into account when performing simulation studies. Several computational docking algorithms have been developed to address these challenges associated with peptides simulations. These can be split into to three classes (Figure 7): template-based docking, local docking and global docking[56]. Their classification is based on the protein-peptide interactions details, whether the structures are known or unknown and the binding site information[56].
|
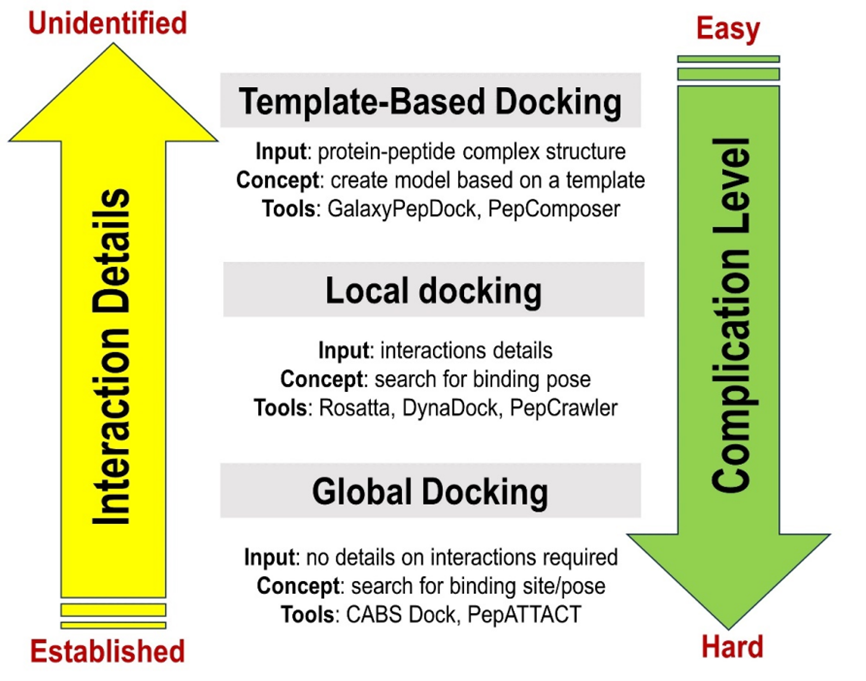
Figure 7. Classification of Protein-Peptide Docking Methods and Some Examples of the Tools Used for Peptide-Protein Docking[56].
3.1.4 Docking Approaches
3.1.4.1 Template-based Docking
The template-based docking is based on the exploitation of known templates -scaffolding proteins- to predict the model protein-peptide complex[21,57,58]. This docking category could be carried out by using a range of tools to analyse sequence-structures manually, semi-automatically or fully automatically. If the investigated complex and the used template are quite similar, this category may be appropriate, successful, and useful[56]. One tool that applies this protocol is GalaxyPepDock webserver which predicts protein-peptide interactions based on templates[57]. It uses protein structure in standard PDB format and a peptide sequence as FASTA format as input files and predicts as output the protein-peptide complex structures[57].
The GalaxyPepDock runs template-based peptide-protein docking in two separate phases[21]. Primarily, the selection of templates by searching in the PepBind Database or PDB which contain structural information for peptide-protein complexes[59], the choosing stage is based on similarities of two scores, the structure of the protein of interest measured by the template modelling score “TM-score”[60,61], and the peptide-protein interactions measured by interactions similarity score “Sinter”. The top ten complex scores are selected as templates if the score is higher than 90%, those selected templates are used for model building. Secondly, the complex structure optimization, for each selected template, 50 models are generated by the model-building tool of the Galaxy template-based modelling based on the GALAXY molecular modelling package[62,63]. Then, only 10 structures are chosen by the GALAXY energy, and they are either exposed or refined by the GalaxyRefine module[64,65].
3.1.4.2 Local Docking
Local docking techniques run docking close to the protein of interest’s known binding active site, however the accuracy of the results depends heavily on the input data. A numerous programs have been developed that require less strictly defined input model, such as Rosetta FlexPepDock. It is one of the first protocols to explicitly include peptide flexibility in docking[66]. DynaDock is another web server for peptide-protein docking that also include receptor flexibility into account[67]. PepCrawler is another tool for high binding affinity of peptide inhibitors that adopts local docking algorithm[68].
3.1.4.3 Global Docking
On the other hand, using the first or second class of computational protein-peptide docking methods to predict complex structures is quite difficult when information on the nature of the binding interactions is not available[69]. In such cases, global docking methods are used in. To dock peptide molecules, this class searches the entire protein surface, so a predefined binding site or peptide-protein complex structure are not required inputs[57]. This search pipeline typically consists of three phases: the prediction of input peptide conformations, docking and model generation, and scoring the models and refinement phase. However, other methods may be used. PEP-SiteFinder[70] and CABS-dock[71-73] are two examples of web servers used for global docking that do not require information about the binding site.
3.1.5 Flexible Peptide, Flexible Protein – A Major Challenge in Docking in General
Structural flexibility of the ligand (a peptide in this case) and target protein is considered the most challenging aspect facing docking simulation at several levels[74]. Protein-peptide complexes are usually formed by involving the folding of the flexible peptide meanwhile binding to the targeted protein[75]. A higher percentage of flexible bonds makes docking operations more difficult and produces unreliable results. Indeed, the number of peptide’s flexible bonds affects directly the prediction accuracy. Taking into account the dynamic nature of proteins, receptor flexibility is identified as an important factor for peptide-protein interactions[58,76]. In fact, during the binding event, protein flexibility may change from side chain bonds to large scale backbone[77]. Numerous methods ignore the receptor’s flexibility in order to reduce computational costs, instead using rigid-body protein docking[56]. For example, CrankPep[78] adopts this approach for the docking of flexible peptides to rigid-body receptors. However, taking receptor flexibility into account directly affects the docking process, leading to inconsistent docking results[79-81].
Numerous flexible peptide-protein docking protocols have been created to address this challenge, and in the text that follows, we highlight the various tools available for flexible peptide-protein docking. Table 1 includes a list of these techniques/tools in summary form. Some tools allows for partially side chain flexibility - with a possibility to include the protein backbone – such as PepCrawler[68], DINC 2.0[82], AutoDock Vina[83-85], Rosetta FlexPepDock[86] Fragments-based docking implemented in PIPER-FlexPepDock[87], is one useful tool for flexible peptide-protein docking that applies global docking algorithm with a high resolution approach modelling of high peptide-protein interactions leading to detailed analyses of theses interactions by using fragment ensembles[87]. To consider the receptor flexibility, there is other class of protocols using soft-core potentials imitators of implicit receptor flexibility, including DynaDock[88]. In addition, CABS-Dock server is used for flexible peptide-protein docking, but it is limited to small backbone fluctuations[71-73]. HADDOCK[89] is one of the tools used for flexible protein-peptide docking. 2 considers the peptide flexibility through an ensemble of peptide conformations generated by our MODPEP program[90].
There are several protocols designed specifically to mimic covalent peptide bonding. One widely used tool is Rosetta CovPepDock which is a high-resolution peptide-protein docking refinement protocol for the modelling of peptide-protein complexes that was developed specifically for covalent flexible peptide[91]. Molsoft incorporates modules for covalent flexible peptides docking[92], Schrodinger CovDock[93], Gold[94], DOCKTITE-MOE[95] and AutoDock4[96], and FITTED[97] are also commonly used for covalent docking, along with other tools that are summarized in Table 1. Comparative evaluation of covalent docking tools is demonstrated in this report[98].
Table 1. Different Tools Used for Flexible Non-Covalent and Covalent Docking and Their Access Links
Peptide-Protein Docking Tool |
Overview |
Link |
Non-Covalent Peptide-Protein Docking |
||
PepCrawler |
Flexible peptide, only protein side chain is flexible. Protein backbone can be made flexible at a computational cost. |
|
DINC 2.0 |
Flexible peptide, only protein side chain is flexible. Protein backbone can be made flexible at a computational cost. |
|
AutoDock Vina |
Flexible peptide, only protein side chain is flexible. Protein backbone can be made flexible at a computational cost. |
|
PIPER-FlexPepDock |
It uses global docking algorithm with a high-resolution approach modelling of high peptide-protein interactions using fragment-based ensembles. |
|
Rosetta FlexPepDock |
Flexible full-atom refinement of PIPER docked models, to generate highly accurate structural models of a peptide-protein interaction. |
|
DynaDock |
Molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. |
Reference[88] |
HPEPDOCK |
Blind peptide-protein docking based on a hierarchical algorithm. |
|
Covalent Peptide-Protein Docking |
||
Rosetta CovPepDock |
Designed for peptide binders that form an irreversible covalent bond with a target cysteine. |
|
Molsoft |
It applies direct stochastic global energy optimization from multiple starting positions of the ligand. |
|
Schrodinger CovDock |
It makes use of data from both the Glide scores of the free pre-reactive species and the Glide score of the final predicted binding mode for covalently bound species. This scoring function captures the fitness of the covalent bond implicitly but does not directly model the bond formation energy. No available information if it is optimized/parametrized for peptide ligands. |
|
Gold |
It consists of conventional noncovalent docking, heuristic formation of the covalent attachment point, and structural refinement of the protein-ligand complex. No information if it is specifically parameterized for peptide ligands. |
|
AutoDock4 |
AutoDock provides quick prediction of bound conformations with predicted free energies of association by combining an empirical free energy force field with a Lamarckian genetic algorithm. No information if it is parameterized for peptides |
|
DOCKTITE-MOE |
It combines automated warhead screening, nucleophilic side chain attachment, pharmacophore-based docking, and a novel consensus scoring approach. It is integrated with MOE software. |
|
FITTED |
FITTED is based on a genetic algorithm (GA) with an emphasis on balancing speed and accuracy. |
|
3.1.6 Sampling and Scoring Function–The Trade-off Between Accuracy and Computational Cost
Sampling and scoring functions are two pillars of each existing docking protocol, However, they are directly related to numerous challenges as they influence the docking result accuracy by a combined effect[76]. Sampling is a crucial docking step because drug discovery requires attention to sampling, speed, and acceleration. There are four major classes of sampling methods classified based on the level of receptor flexibility: (1) The simplest category, soft docking, treats implicitly only a portion of target flexibility (cannot be used for large side chain or backbone); (2) selective docking is based on choosing only a few crucial residues that are linked to the peptide receptors. This approach might work if the receptor’s structural information is available; (3) ensemble docking usually considers full flexibility of the receptor implicitly; (4) on-the-fly docking considers receptor flexibility explicitly as it generates new receptor conformations on the fly during the docking process, however, treating receptor and peptide flexibility simultaneously increase the complexity of the operation. Detailed information on sampling techniques can be found in this source[76].
On the other hand, the scoring functions also contributes to the accuracy of docking results. scoring function is a ranking system consists of binding interaction estimation (binding energy) for a specific bond[99]. In fact, for flexible protein-peptide docking methods, scoring function takes the position of every peptide atom and each protein atom and converts it to a numeric value as a score, this is the procedure to rank the potential conformations of docking results, toward to select the most reasonable conformations to symbolize flexible peptide-protein complex in vivo[100]. However, to identify the highest accurate complex from all docking results is the challenge in this operation[56].
In addition, the crucial difficulty facing docking approaches with covalent peptides is the formation of covalent bonds between the electrophile peptide’s functional groups and the nucleophilic residue in the active site of a receptor. The biggest portion of docking programs specific to covalent docking are incapable to perform an accurate estimation of covalent binding energy, because the covalent bond formation energy is excluded from the general score. In general, three classes of scoring methods can be distinguished: (1) force-fields based scoring functions; (2) empirical or semi empirical scoring functions; (3) knowledge-based functions that are elaborated by using statistical data of well-known structures of peptide-protein complex[101]. CAPRI is communitywide experiment on the comparative evaluation of protein-protein docking for structure prediction demonstrated that the best peptide-protein docking results are generated by using methods established on mixed scoring functions[58,102].
Finally, integrating experimental data into computational calculations has been implemented in various docking scoring functions and algorithms in order to improve docking accuracy. A number of docking programs have included cryo-EM data into their operational procedures. For example MultiFit automatically segments the cryo-EM density using a Gaussian mixture model to deduce anchors, subsequently docking the components of the complex onto the anchors[103]. Using 3D Zernike descriptors, EMLZerD scores the models based on the cryo-EM data[104]. A recent method has been used in ATTRACT-EM that fits the subunits into the map using a Gaussian mixture model to represent the cryo-EM data, and then refines the resulting models[100]. However, it is challenging to convert the experimental data into computational information of the covalent peptide-protein interactions during the docking process. Apparently, these approach encounters a lot of difficulties and ambiguities in terms of the conversion robustness of the available experimental data[101]. Most of these methods separate the use of the cryo-EM data from the use of other sources of information. Some important aspects such as physico-chemical properties (energetics) are only taken into consideration after fitting or refining structural parameters. Additionally, they typically do not make active use of any additional orthogonal data that might be provided, like mass spectrometry cross-link data or mutagenesis data. HADDOCK is one useful tool that integrates Cryo-EM and other experimental data in its docking algorithm to enhance docking scoring functionality[89,105].
3.1.7 Flexible Protein-peptide Docking Methods – A Way Forward
Simulations of covalent inhibitor binding remain a challenging topic, with many technical considerations to be taken into account. The starting point of performing covalent binding simulations is covalent docking. Although molecular docking methods in general have undergone significant development over time, covalent flexible docking, especially for peptides, remains a huge technical challenge. Docking accuracy depends on many factors, such as the size and architecture of peptide-protein complex, the, sampling method, the choice of parameters and scoring functions and the flexibility of both the protein and peptide. Peptides conformational flexibility and folding motif present another worrying challenge. Besides, there is no unique protocol or specific criteria that could be possibly used to obtain the best results. Another obstacle might be the protein low-resolution crystal structure or a missing amino acid sequence. Lack of information on binding sites adds another difficulty to the process. Overall, the accuracy of predicting a peptide-protein complexes is the main object, and the major difficulty for any predictor is modelling the bound conformation of the protein-peptide interactions with best accuracy levels.
We believe that a concise technical guide on each challenge and a list of potential solutions would be helpful to researchers working on improving docking protocols (Table 2). These guidelines are primarily based on our experience we have accumulated over the years in covalent docking research[23-29].
Table 2. Challenges of Flexible Peptide Covalent Docking and Potential Solutions and Intervention
Challenge |
Possible Solutions |
Unavailability or low resolution of protein crystal structure |
Homology modelling approach can be used to address this challenge. A few technical tips on homology modelling of protein structure are provided in refs[106,107]. AlphaFold Artificial Intelligence (AlphaFold AI) is another useful tool for prediction of protein 3D structures[108]. |
Missing residues in the protein structure |
There are several tools to address this challenge. SL2 is one of the easy-to-use and efficient tools that can be used. It is a fragment-based tool for the prediction and interactive placement of loop structures into globular and helical membrane proteins[109]. |
No available information on the binding site |
Binding site prediction tools can be used. A wide range of tools that can be used for this purpose and validations protocols are available. See provided references[110,111]. |
Flexibility/conformations of peptides |
It is essential to determine the lowest energy conformations of peptides and most importantly their folding motif[112]. A few techniques have been developed for modelling protein-bound peptide conformations, Here we present a list of tools that can be used to predict peptide structures and folding motif: MODPEP can be used for conformational sampling of protein-bound peptides[113]. PEP-FOLD (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD/), APPTEST (https://research.timmons.eu/apptest_help) and PEPstrMOD (https://webs.iiitd.edu.in/raghava/pepstrmod/) are useful tools for predication peptide structures.
Two review articles tackle problems related to peptides such as predicting structures, binding affinities and even kinetics[112,114] would also serve as useful resources in this regard.
Performing molecular dynamics simulations of covalent bonding is technically challenging, however, we recently published a detailed technical guide on how to perform all-molecular-mechanics MD for covalent simulations[115]. |
Flexible peptide, flexible protein |
Although there are several docking tools that take flexibility of both protein and peptide into account (listed in Table 1), it is strongly recommended that molecular dynamics simulations can be used in conjunction with docking calculations to verify docking results. A few published protocols can be accessed here[116-118]. |
Too many scoring functions and algorithms-which one to pick? |
There is no typical protocol/algorithm that can guarantee the accuracy of docking calculations, hence it is essential to validate docking results using one or more approaches described below. Some useful reports on the covalent docking protocol selection are provided[98,119,120]. |
Validation of docking protocol - a must to do |
There is no doubt that validation of docking results against experimental data is the most acceptable approach, however, several in silico validation protocols can be adopted: Cross-validation by using more than one covalent docking tool to cross-validate the results. Re-docking of experimentally determined X-ray crystal structure of peptide-protein complexes to assess the accuracy of docking in predicting the native binding pose. However, careful attention should be paid here as re-docking can be an artificial exercise. Incorporation of molecular dynamics simulations and thermodynamic calculations such as MM-PBSA/MM-GBSA[121] to estimate the binding affinities. Ensemble docking is a method that is used to generate an “ensemble” of drug target conformations, frequently through the use of molecular dynamics simulation. |
All in all, it is expected that the forthcoming protocols would be able to combine more than one approach, providing facilities to integrate experimental and computational docking results. The advances in computational performance and the integration of Artificial Intelligence and Machine learning would improve the accuracy of docking calculations. The molecular docking process, however, is a multi-dimensional one that calls for a logical protocol, design, and validations-most importantly, against experimental data or counterparts.
4 CONCLUSION
Electrophilic peptides that can attach irreversibly through a covalent bond with target protein offer considerable potential for drug discovery especially for protein targets that were previously thought to be undruggable. Nevertheless, the discovery of such covalent peptides still poses several challenge, hence efficient de novo rational design approaches for covalent-based peptide inhibitors are critical. In this context, herein, we offered a “one stop shop” for in silico rational design of covalent-based inhibitors, associated challenges and possible interventions and expert opinion. This review discussed covalent docking of peptide-based molecules from a technical point of view, as well as a number of theoretical facets that, all together, provide a broad yet crucial knowledge of the principles of rational design of peptide-based inhibitors. The various aspects of peptide covalent docking and flexible peptide-protein docking were also highlighted in this review. Determination of the optimal amino acid sequence serves as the initial yet most crucial starting point of design of peptide-based inhibitors. The peptide sequence can be proposed based on chemical intuition, experimental data, existing peptide drug template, quantitative structure activity relationship, the binding site architecture of target protein, protein-protein interface, etc. It is also crucial to consider that the amino acid sequence of a peptide determines its physicochemical characteristics, toxicity profile, pharmacodynamic landscape, and capacity to bind to a protein target. Because they don’t have electrophilic moieties, natural peptides can’t bind covalently to target proteins, therefore, electrophilic warhead must be incorporated in the structure of the peptide for covalent binding to occur. For this, we showcased the available in silico and bioinformatics tools to assist with the de novo design of peptide sequence, selecting electrophilic warhead and the most reliable tools for covalent docking, taking into account peptide flexibility and folding. We also presented the major challenges of flexible peptide covalent docking and provide potential practical solutions and intervention based on our experience in the field.
We believe that this report adds significantly to the understanding of rational design of peptide-based covalent inhibitors and their applications in drug development and discovery.
Acknowledgements
Not applicable.
Conflicts of Interest
The authors declared no conflict of interest.
Data Availability
No additional data are available.
Copyright Permissions
Copyright © 2024 The Author(s). Published by Innovation Forever Publishing Group Limited. This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.
Author Contribution
Soliman MES conceptualized project idea and set out the project design; Lafifi Z collected data, literature survey and writing up; Soliman MES and Bjij I revised the manuscript and checked all information.
Abbreviation List
PPI, Proton pump inhibitor
References
[1] Ghosh AK, Samanta I, Mondal A et al. Covalent Inhibition in Drug Discovery. ChemMedChem. 2019; 14: 889-906.[DOI]
[2] Sutanto F, Konstantinidou M, Dömling A. Covalent inhibitors: A rational approach to drug discovery. RSC Med Chem, 2020; 11: 876-884.[DOI]
[3] Shin JM, Munson K, Vagin O et al. The gastric HK-ATPase: Structure, function, and inhibition. Pflug Arch Eur J Phy, 2009; 457: 609-622.[DOI]
[4] Munson K, Garcia R, Sachs G. Inhibitor and ion binding sites on the gastric H, K-ATPase. Biochemistry-Us, 2005; 44: 5267-5284.[DOI]
[5] Patti G, Micieli G, Cimminiello C et al. The role of clopidogrel in 2020: A reappraisal. Cardiovasc Ther, 2020; 2020: 8703627.[DOI]
[6] Michalopoulos AS, Livaditis IG, Gougoutas V. The revival of fosfomycin. Int J Infect Dis, 2011; 15:e732-9.[DOI]
[7] Tam CS, Muñoz JL, Seymour JF et al. Zanubrutinib: Past, present, and future. Blood Cancer J, 2023; 13: 141.[DOI]
[8] Lavacchi D, Mazzoni F, Giaccone G. Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): Current perspectives. Drug Des Dev Ther, 2019; 13: 3187-3198.[DOI]
[9] Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science, 1995; 267: 383-386.[DOI]
[10] London N, Movshovitz-Attias D, Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure, 2010; 18: 188-199.[DOI]
[11] Fosgerau K, Hoffmann T. Peptide therapeutics: Current status and future directions. Drug Discov Today, 2015; 20: 122-128.[DOI]
[12] Quianzon CC, Cheikh I. History of insulin. J Community Hosp Int, 2012; 2: 18701.[DOI]
[13] Carter CS. Close encounters with oxytocin. Compr Psychoneuroendocrinol, 2023; 15: 100189.[DOI]
[14] Mitchell AR. Bruce Merrifield and solid‐phase peptide synthesis: A historical assessment. Peptide Sci, 2008; 90: 175-184.[DOI]
[15] Culf AS. Peptoids as tools and sensors. Biopolymers, 2019; 110: e23285.[DOI]
[16] Liu M, Li X, Xie Z et al. D‐peptides as recognition molecules and therapeutic agents. Chem Rec, 2016; 16: 1772-1786.[DOI]
[17] Zorzi A, Deyle K, Heinis C. Cyclic peptide therapeutics: Past, present and future. Curr Opin Chem Biol, 2017; 38: 24-29.[DOI]
[18] Potashman MH, Duggan ME. Covalent modifiers: An orthogonal approach to drug design. J Med Chem, 2009; 52: 1231-1246.[DOI]
[19] Singh J, Petter RC, Baillie TA et al. The resurgence of covalent drugs. Nat Rev Drug Discov, 2011; 10: 307-317.[DOI]
[20] London N, Lamphear CL, Hougland JL et al. Identification of a novel class of farnesylation targets by structure-based modeling of binding specificity. PLoS Comput Biol, 2011; 7: e1002170.[DOI]
[21] Lee H, Heo L, Lee MS et al. GalaxyPepDock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res, 2015; 43: W431-W435.[DOI]
[22] Raveh B, London N, Schueler-Furman O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins, 2010; 78: 2029-2040.[DOI]
[23] Aljoundi A, El Rashedy A, Soliman MES. Comparison of irreversible inhibition targeting HSP72 protein: The resurgence of covalent drug developments. Mol Simulat, 2021; 47: 1093-1103.[DOI]
[24] Issahaku AR, Soliman MES. Investigating The Impact of Covalent and Non-covalent Binding Modes of Inhibitors on Bruton’s Tyrosine Kinase in the Treatment of B Cell Malignancies-Computational Insights. Curr Pharm Biotechno, 2022; 24: 814-824.[DOI]
[25] Bjij I, Ramharack P, Khan S et al. Tracing potential covalent inhibitors of an E3 ubiquitin ligase through target-focused modelling. Molecules, 2019; 24: 3125.[DOI]
[26] Kumalo HM, Bhakat S, Soliman MES. Theory and applications of covalent docking in drug discovery: Merits and pitfalls. Molecules, 2015; 20: 1984-2000.[DOI]
[27] Aljoundi A, Bjij I, El Rashedy A et al. Covalent Versus Non-covalent Enzyme Inhibition: Which Route Should We Take? A Justification of the Good and Bad from Molecular Modelling Perspective. Protein J, 2020; 39: 97-105.[DOI]
[28] Khan S, Bjij I, Soliman MES. Selective Covalent Inhibition of “Allosteric Cys121” Distort the Binding of PTP1B Enzyme: A Novel Therapeutic Approach for Cancer Treatment. Cell Biochem Biophys, 2019; 77: 203-211.[DOI]
[29] Bjij I, Khan S, Ramharak P et al. Distinguishing the optimal binding mechanism of an E3 ubiquitin ligase: Covalent versus noncovalent inhibition. J Cell Biochem, 2019; 120: 12859-12869.[DOI]
[30] Ghosh AK, Samanta I, Mondal A et al. Covalent Inhibition in Drug Discovery. Vol. 14, ChemMedChem, 2019; 14: 889–906.[DOI]
[31] Flanagan ME, Abramite JA, Anderson DP et al. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J Med Chem, 2014; 57: 10072-10079.[DOI]
[32] Aljoundi A, Bjij I, El Rashedy A et al. Covalent Versus Non-covalent Enzyme Inhibition: Which Route Should We Take? A Justification of the Good and Bad from Molecular Modelling Perspective. Protein J, 2020; 39: 97-105.[DOI]
[33] Bjij I, Olotu FA, Agoni C et al. Covalent Inhibition in Drug Discovery: Filling the Void in Literature. Curr Top Med Chem. 2018; 18: 1135–1145.[DOI]
[34] Wang ML, Jurczak W, Zinzani PL et al. Pirtobrutinib in Covalent Bruton Tyrosine Kinase Inhibitor Pretreated Mantle-Cell Lymphoma. J Clin Oncol, 2023; 41: 3988–3997.[DOI]
[35] Mato AR, Woyach JA, Brown JR et al. Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia. N Engl J Med, 2023; 389: 33–44.
[36] Rhodes JM, Mato AR. Zanubrutinib (BGB-3111), a Second-Generation Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase and Its Utility in Treating Chronic Lymphocytic Leukemia. Drug Des Devel Ther, 2021; 15: 919–26.[DOI]
[37] Yang T, Cuesta A, Wan X et al. Reversible lysine-targeted probes reveal residence time-based kinase selectivity. Nat Chem Biol, 2022; 18: 934-941.[DOI]
[38] Devkota AK, Edupuganti R, Yan C et al. Reversible covalent inhibition of eEF-2K by carbonitriles. ChemBioChem, 2014; 15: 2435-2442.[DOI]
[39] Liu M, Xu B, Ma Y et al. Reversible covalent inhibitors suppress enterovirus 71 infection by targeting the 3C protease. Antiviral Res, 2021; 192: 105102.[DOI]
[40] Awoonor-Williams E. Estimating the binding energetics of reversible covalent inhibitors of the SARS-CoV-2 main protease: An in silico study. Phys Chem Chem Phys, 2022; 24: 23391-23401.[DOI]
[41] Liang C, Tian D, Ren X et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur J Med Chem, 2018;151: 315-326.[DOI]
[42] Zhang H, Jiang W, Chatterjee P et al. Ranking Reversible Covalent Drugs: From Free Energy Perturbation to Fragment Docking. J Chem Inf Model, 2019; 59: 2093-2102.[DOI]
[43] Smith S, Keul M, Engel J et al. Characterization of Covalent-Reversible EGFR Inhibitors. ACS Omega, 2017; 2: 1563-1575.[DOI]
[44] Serafimova IM, Pufall MA, Krishnan S et al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol, 2012; 8: 471-476.[DOI]
[45] Pearson RG, Dillon RL. Rates of Ionization of Pseudo Acids.1 IV. Relation between Rates and Equilibria. J Am Chem Soc, 1953; 75: 2439-2443.[DOI]
[46] Sogabe S, Kawakita Y, Igaki S et al. Structure-based approach for the discovery of pyrrolo [3, 2-d] pyrimidine-based EGFR T790M/L858R mutant inhibitors. ACS Med Chem Lett, 2012; 4: 201-205.[DOI]
[47] Lee MF, Poh CL. Strategies to improve the physicochemical properties of peptide-based drugs. Pharm Res, 2023; 40: 617-632.[DOI]
[48] D’Annessa I, Di Leva FS, La Teana A et al. Bioinformatics and Biosimulations as Toolbox for Peptides and Peptidomimetics Design: Where Are We?. Front Mol Biosci, 2020; 7: 66.[DOI]
[49] Zheng L, Li Y, Wu D et al. Development of covalent inhibitors: Principle, design, and application in cancer. MedComm-Oncol, 2023; 2: e56.[DOI]
[50] Péczka N, Orgován Z, Ábrányi-Balogh P et al. Electrophilic warheads in covalent drug discovery: An overview. Expert Opin Drug Discov, 2022; 17: 413-422.[DOI]
[51] Paulussen FM, Grossmann TN. Peptide‐based covalent inhibitors of protein-protein interactions. J Pept Sci, 2023; 29: e3457.[DOI]
[52] Assaf L, Chi Ho N, Dana M et al. Detection of peptide‐binding sites on protein surfaces: The first step toward the modeling and targeting of peptide‐mediated interactions. Proteins, 2014; 82: 1550-1550.[DOI]
[53] Compain G, Monsarrat C, Blagojevic J et al. Peptide-Based Covalent Inhibitors Bearing Mild Electrophiles to Target a Conserved His Residue of the Bacterial Sliding Clamp. JACS Au, 2024; 4: 432-440.[DOI]
[54] Tivon B, Gabizon R, Somsen BA et al. Covalent flexible peptide docking in Rosetta. Chem Sci, 2021 ;12: 10836-10847.[DOI]
[55] Ho BK, Dill KA. Folding Very Short Peptides Using Molecular Dynamics. PLoS Comput Biol, 2006; 2: e27.[DOI]
[56] Ciemny M, Kurcinski M, Kamel K et al. Protein-peptide docking: opportunities and challenges. Drug Discov Today, 2018; 23: 1530-1537.[DOI]
[57] Lee H, Seok C. Template-based prediction of protein-peptide interactions by using GalaxyPepDock. Model Peptide-Protein Interact: Method Protocol, 2017; 1561: 37-47.[DOI]
[58] Lensink MF, Velankar S, Wodak SJ. Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins, 2017; 85: 359-377.[DOI]
[59] Das AA, Sharma OP, Kumar MS et al. PepBind: A comprehensive database and computational tool for analysis of protein-peptide interactions. Genom Proteom Bioinf, 2013; 11: 241-246.[DOI]
[60] Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins, 2004; 57: 702-710.[DOI]
[61] Zhang Y, Skolnick J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res, 2005; 33: 2302-2309.[DOI]
[62] Park H, Ko J, Joo K et al. Refinement of protein termini in template‐based modeling using conformational space annealing. Proteins, 2011; 79: 2725-2734.[DOI]
[63] Ko J, Park H, Seok C. GalaxyTBM: Template-based modeling by building a reliable core and refining unreliable local regions. BMC Bioinformatics, 2012; 13: 1-8.[DOI]
[64] Heo L, Park H, Seok C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res, 2013; 41: W384-W388.[DOI]
[65] Heo L, Lee H, Seok C. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci Rep, 2016; 6: 32153.[DOI]
[66] Marcu O, Dodson EJ, Alam N et al. FlexPepDock lessons from CAPRI peptide-protein rounds and suggested new criteria for assessment of model quality and utility. Proteins, 2017; 85: 445-462.[DOI]
[67] Antes I. DynaDock: A new molecular dynamics‐based algorithm for protein-peptide docking including receptor flexibility. Proteins, 2010; 78: 1084-1104.[DOI]
[68] Donsky E, Wolfson HJ. PepCrawler: A fast RRT-based algorithm for high-resolution refinement and binding affinity estimation of peptide inhibitors. Bioinformatics, 2011; 27: 2836-2842.[DOI]
[69] Lee H, Heo L, Lee MS et al. GalaxyPepDock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res, 2015; 43: W431-W435.[DOI]
[70] Saladin A, Rey J, Thévenet P et al. PEP-SiteFinder: A tool for the blind identification of peptide binding sites on protein surfaces. Nucleic Acids Res, 2014; 42: W221-W226.[DOI]
[71] Kurcinski M, Badaczewska‐Dawid A, Kolinski M et al. Flexible docking of peptides to proteins using CABS‐dock. Protein Sci, 2020; 29: 211-222.[DOI]
[72] Kurcinski M, Jamroz M, Blaszczyk M et al. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res, 2015; 43: W419-W424.[DOI]
[73] Kurcinski M, Blaszczyk M, Ciemny MP et al. A protocol for CABS-dock protein-peptide docking driven by side-chain contact information. Biomed Eng Online, 2017; 16: 1-10.[DOI]
[74] Bonvin AMJJ. Flexible protein-protein docking. Curr Opin Struc Biol, 2006; 16: 194-200.[DOI]
[75] Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struc Biol, 2009; 19: 31-38.[DOI]
[76] Antunes DA, Devaurs D, Kavraki LE. Understanding the challenges of protein flexibility in drug design. Expert Opin Drug Dis, 2015; 10: 1301-1313.[DOI]
[77] Buonfiglio R, Recanatini M, Masetti M. Protein flexibility in drug discovery: From theory to computation. ChemMedChem, 2015; 10: 1141-1148.[DOI]
[78] Zhang Y, Sanner MF. AutoDock CrankPep: Combining folding and docking to predict protein-peptide complexes. Bioinformatics, 2019; 35: 5121-5127.[DOI]
[79] Ding F, Dokholyan NV. Incorporating backbone flexibility in MedusaDock improves ligand-binding pose prediction in the CSAR2011 docking benchmark. J Chem Inf Model, 2013; 53: 1871-1879.[DOI]
[80] Feixas F, Lindert S, Sinko W et al. Exploring the role of receptor flexibility in structure-based drug discovery. Biophys Chem, 2014; 186: 31-45.[DOI]
[81] Sinko W, Lindert S, Mccammon JA. Accounting for Receptor Flexibility and Enhanced Sampling Methods in Computer-Aided Drug Design. Chem Biol Drug Des. 2013; 81: 41-49.[DOI]
[82] Antunes DA, Moll M, Devaurs D et al. DINC 2.0: A new protein-peptide docking webserver using an incremental approach. Cancer Res, 2017; 77: e55-e57.[DOI]
[83] Wang Z, Sun H, Yao X et al. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys Chem Chem Phys, 2016; 18: 12964-12975.[DOI]
[84] Abreu RMV, Froufe HJC, Queiroz MJRP et al. Selective flexibility of side‐chain residues improves VEGFR‐2 docking score using AutoDock Vina. Chem Biol Drug Des, 2012; 79: 530-534.[DOI]
[85] Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem, 2010;31: 455–461.[DOI]
[86] London N, Raveh B, Cohen E et al. Rosetta FlexPepDock web server-high resolution modeling of peptide-protein interactions. Nucleic Acids Res, 2011; 39: W249-W253.[DOI]
[87] Alam N, Goldstein O, Xia B et al. High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock. PLoS Comput Biol, 2017; 13: e1005905.[DOI]
[88] Antes I. DynaDock: A new molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins. 2010; 78: 1084–104.[DOI]
[89] Geng C, Narasimhan S, Rodrigues JPGLM et al. Information-Driven, Ensemble Flexible Peptide Docking Using HADDOCK. Methods Mol Biol. 2017; 1561: 109–138.[DOI]
[90] Zhou P, Jin B, Li H et al. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res, 2018; 46: W443-W450.[DOI]
[91] Tivon B, Gabizon R, Somsen BA et al. Covalent flexible peptide docking in Rosetta. Chem Sci, 2021; 12: 10836-10847.[DOI]
[92] Fernández‐Recio J, Totrov M, Abagyan R. ICM‐DISCO docking by global energy optimization with fully flexible side‐chains. Proteins, 2003; 52: 113-117.[DOI]
[93] Zhu K, Borrelli KW, Greenwood JR et al. Docking covalent inhibitors: A parameter free approach to pose prediction and scoring. J Chem Inf Model, 2014; 54: 1932-1940.[DOI]
[94] David L, Mdahoma A, Singh N et al. A toolkit for covalent docking with GOLD: From automated ligand preparation with KNIME to bound protein-ligand complexes. Bioinform Adv, 2022; 2: vbac090.[DOI]
[95] Scholz C, Knorr S, Hamacher K et al. DOCKTITE–A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment. J Chem Inf Model, 2015; 55: 398-406.[DOI]
[96] Morris GM, Huey R, Lindstrom W et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem, 2009; 30: 2785-2791.[DOI]
[97] Moitessier N, Pottel J, Therrien E et al. Medicinal chemistry projects requiring imaginative structure-based drug design methods. Accounts Chem Res, 2016; 49: 1646-1657.[DOI]
[98] Scarpino A, Ferenczy GG, Keserű GM. Comparative evaluation of covalent docking tools. J Chem Inf Model, 2018; 58: 1441-1458.[DOI]
[99] Grinter SZ, Zou X. Challenges, applications, and recent advances of protein-ligand docking in structure-based drug design. Molecules, 2014; 19: 10150-10176.[DOI]
[100] Dhanik A, Kavraki LE. Protein-ligand interactions: Computational docking. eLS, 2012; 2012: 15.[DOI]
[101] Sotriffer C. Docking of covalent ligands: Challenges and approaches. Mol Inform, 2018; 37: e1800062.[DOI]
[102] Yu J, Andreani J, Ochsenbein F et al. Lessons from (co-)evolution in the docking of proteins and peptides for CAPRI Rounds 28-35. Proteins, 2017; 85: 378–390.[DOI]
[103] Lasker K, Sali A, Wolfson HJ. Determining macromolecular assembly structures by molecular docking and fitting into an electron density map. Proteins, 2010; 78: 3205-3211.[DOI]
[104] Esquivel-Rodríguez J, Kihara D. Fitting multimeric protein complexes into electron microscopy maps using 3D Zernike descriptors. J Phys Chem, 2012; 116: 6854-6861.[DOI]
[105] Deplazes E, Davies J, Bonvin AMJJ et al. Combination of ambiguous and unambiguous data in the restraint-driven docking of flexible peptides with HADDOCK: The binding of the spider toxin PcTx1 to the acid sensing ion channel (ASIC) 1a. J Chem Inf Model, 2016; 56: 127-138.[DOI]
[106] França TCC. Homology modeling: An important tool for the drug discovery. J Biomol Struct Dyn, 2015; 33: 1780-1793.[DOI]
[107] Muhammed MT, Aki‐Yalcin E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem Biol Drug Des, 2019; 93: 12-20.[DOI]
[108] Varadi M, Anyango S, Deshpande M et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res, 2022; 50: D439-D444.[DOI]
[109] Ismer J, Rose AS, Tiemann JKS et al. SL2: An interactive webtool for modeling of missing segments in proteins. Nucleic Acids Res, 2016; 44: W390-W394.[DOI]
[110] Broomhead NK, Soliman ME. Can we rely on computational predictions to correctly identify ligand binding sites on novel protein drug targets? Assessment of binding site prediction methods and a protocol for validation of predicted binding sites. Cell Biochem Biophys, 2017; 75: 15-23.[DOI]
[111] Xie ZR, Hwang MJ. Methods for predicting protein-ligand binding sites. Mol Model Proteins, 2014; 2014: 383-398.[DOI]
[112] London N, Raveh B, Schueler-Furman O. Peptide docking and structure-based characterization of peptide binding: From knowledge to know-how. Curr Opin Struc Biol, 2013; 23: 894-902.[DOI]
[113] Yan Y, Zhang DI, Huang SY. Efficient conformational ensemble generation of protein-bound peptides. J Cheminformatics, 2017; 9: 1-13.[DOI]
[114] Mondal A, Chang L, Perez A. Modelling peptide-protein complexes: Docking, simulations and machine learning. QRB Discov, 2022; 3: e17.[DOI]
[115] Khan S, Bjij I, Olotu F A et al. Covalent simulations of covalent/irreversible enzyme inhibition in drug discovery: A reliable technical protocol. Future Med Chem, 2018; 10: 2265-2275.[DOI]
[116] Naqvi AAT, Mohammad T, Hasan GM et al. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr Top Med Chem, 2018; 18: 1755-1768.[DOI]
[117] Santos LHS, Ferreira RS, Caffarena ER. Integrating molecular docking and molecular dynamics simulations. Docking Scre Drug Discov, 2019; 2019: 13-34.[DOI]
[118] Gioia D, Bertazzo M, Recanatini M et al. Dynamic docking: A paradigm shift in computational drug discovery. Molecules, 2017; 22: 2029.[DOI]
[119] Sotriffer C. Docking of covalent ligands: Challenges and approaches. Mol Inform, 2018; 37: 1800062.[DOI]
[120] Hevener KE, Zhao W, Ball DM et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model, 2009; 49: 444-460.[DOI]
[121] Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Dis, 2015; 10: 449-461.[DOI]
Brief of Corresponding Author(s)
Mahmoud E.S. Soliman He is a Full Professor at the Department of Pharmaceutical Sciences, School of Health Sciences, UKZN, Westville Campus, and the Head and Principal Investigator of Molecular Bio-Computation and Drug Design Laboratory. He completed his postgraduate degree (MPhil/PhD - 2009) at the University of Bath, United Kingdom. Prof. Soliman’s research mainly focuses on studying biomolecular systems, drug-protein interactions, and rational design of drug candidates using experimental and computational techniques. He has published more than 320 publications in internationally accredited journals. Soliman accumulated H-index of 37. |



