From Biological Activity to Stereoselectivity: A Portrait of Molecular and Mechanistic Profiles of the Therapeutic Potential of G-1 and LNS8801 as GPER-1 Activator in the Treatment of Waldenström’s Macroglobulinemia
Ghazi Elamin1,2 Aimen Aljoundi1,3, Mahmoud E.S. Soliman1*![]()
1Molecular Bio-computation and Drug Design Laboratory School of Health Sciences, University of KwaZulu-Natal, Westville Campus, Durban, South Africa
2Department of Pharmaceutical Chemistry, Collage of Pharmacy, Karary University, Khartoum, Sudan
3Department of Pharmaceutical Chemistry, Collage of Pharmacy, University of Attahadi, Tripoli, Libya
*Correspondence to: Mahmoud E.S. Soliman, PhD, Professor, Molecular Bio-computation and Drug Design Laboratory School of Health Sciences, University of KwaZulu-Natal, Westville Campus, Durban, 4001, South Africa; Email: soliman@ukzn.ac.za
DOI: 10.53964/id.2024007
Abstract
Keywords: GPER-1, Waldenström's Macroglobulinemia, G-1, LNS8801, enantiomer
1 INTRODUCTION
Waldenström’s Macroglobulinemia (WM) is an incurable B-cell malignancy characterized by the presence of monoclonal immunoglobulin M (IgM) in the bloodstream and the infiltration of clonal lymphoplasmacytic cells in the bone marrow[1]. It accounts for approximately 1% to 2% of all hematologic malignancies, with an estimated annual incidence of 1500 new cases in the United States[2-4]. The average survival rate for symptomatic individuals is approximately nine years[5]. The exact cause of WM is unknown, but it is believed to result from genetic mutations that lead to uncontrolled growth and accumulation of lymphoplasmacytic cells[6]. The abnormal cells produce excessive amounts of monoclonal IgM, a large antibody molecule, which can cause various symptoms and complications[7,8] Standard treatment for WM involves the administration of rituximab, an anti-CD20 antibody, in combination with alkylating agents, nucleoside analogues, and proteasome antagonists. In addition, novel therapeutic approaches, such as next-generation covalent BTK inhibitors, non-covalent BTK inhibitors, BCL-2 inhibitors, and CXCR4-targeted drugs, are being explored[9-12]. However, the current standard of care only achieves complete remission in a small percentage of WM patients, underscoring the urgent need for the development of new therapeutic strategies. The G protein–coupled receptor (GPCR) superfamily is the largest and most diverse cell surface receptors involved in many critical physiological processes, diseases, and disorders[13]. Moreover, while at least 500 GPCRs have been revealed as therapeutically significant targets, only a small fraction of GPCRs have been described either in their free apo form or in the presence of ligands[14]. G protein-coupled estrogen receptor 1 (GPER-1/GPR30) is a member of the G protein-coupled receptor that controls cellular processes in cancer, including proliferation, migration, and apoptosis[15-19]. GPER-1 has garnered attention as a promising and innovative therapeutic target based on its role in facilitating the transactivation of the epidermal growth factor receptor (EGFR), activation of the MAPK and PI3K signalling pathways, stimulation of adenyl cyclase, and mobilization of intracellular calcium[20]. The pharmacological activation of GPER-1 with the selective small-molecule agonist G-1 or its enantiomer LNS8801 is one of the promising new approaches to treating WM and other human cancers[21-23]. G-1 and LNS8801 induce an increase in mitotic protein cyclin B1 expression, as well as annexin V binding and caspase 3/7 activity, two indicators of apoptotic cell death and the accumulation of BCWM-1 cells in the G2/M phase[23].
Enantiomers, or optical isomers, are chiral molecules with different three-dimensional (3D) geometries and no axis of symmetry but identical atomic sequences and bonds[24]. Although enantiomers share similar physical characteristics, like melting and boiling temperatures, their biological activities and mechanisms may differ[24]. Enantiomers whose plane of polarization rotates counter-clockwise are known as laevorotary or negative (l or -), whereas those whose plane rotates clockwise are known as dextrorotary or positive (d or +)[24]. The racemic mixture contains both the positive and negative enantiomers in equal amounts[24,25]. The administration of racemic drugs boosts enantiomer-enantiomer interactions, with one enantiomer vying with the other for binding to a receptor's active region[26]. Stereoselective interactions can happen as one enantiomer may have more potent interactions, leading to preferential binding[24,25]. These pharmacological activity differences provide important clues about the medications’ mechanisms of action and may be valuable in fine-tuning the enantiomer ratio for future research into the potential therapeutic benefits of enantiomers in therapeutic interventions[27,28]. Recent research has shown that pharmacologically activating GPER-1 suppresses tumor cell proliferation in WM. G-1 (Tespria) and LNS8801, a selective small-molecule agonist of GPER-1, causes G2/M cell cycle arrest and apoptosis in vitro and in vivo in animal models[23].
Biomedical engineers are striving to develop simple, inexpensive, portable, and powerful tools that might be useful in numerous fields of biomedical research, including drug discovery and therapeutic methods in regenerative medicine. This is the aim of biomaterials research and development, which seeks to create low-cost, broadly applicable technologies[29,30]. Computer-aided drug design (CADD) is extensively used to expedite the discovery of effective in-vitro and in-vivo models and identify small molecules with inhibitory potential[31,32]. This study focused on using computational methods, specifically MD modeling, on investigating the structural dynamics of GPER-1 following stereoselective interactions with G-1 and its enantiomer LNS8801 (Figure 1). Using several post-MD molecular modeling analysis approaches, we gained structural insight into the binding abilities of G-1, its enantiomer LNS8801, and the conformational changes that occurred to the GPER-1 enzyme following binding. This report’s findings will inform future efforts to enhance and optimize these hits to increase their potency and selectivity as potential drug candidates.
|
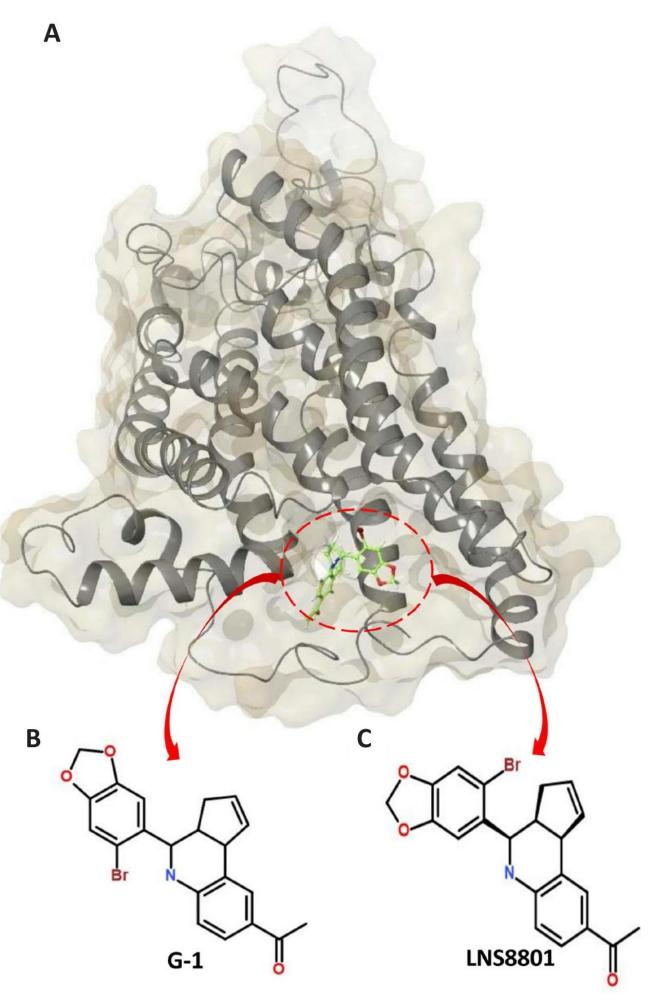
Figure 1. The Structural Dynamics of GPER-1 Following Stereoselective Interactions with G-1 and its Enantiomer LNS8801. A: Surface view of the GPER-1 enzyme bound to G-1 and LNS8801.B: 2D structures of G-1. C: 2D structures of LNS8801.
2 COMPUTATIONAL METHODOLOGY
2.1 System Preparation
AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/)[33,34], was used to predict the 3D structural of G protein-coupled estrogen receptor 1 (GPER-1) using FASTA sequence which obtained from UniProt (https://www.uniprot.org/) (UniProt ID: Q99527)[35]. Structure of GPER-1 was prepared for molecular dynamics simulation using Molegro Molecular Viewer[36], and the UCSF Chimera software tool[37]. The SDF files for G-1 (ID: 3136849) and LNS8801 (ID: 5322399) were downloaded from Pubchem[38], and used the web service Open Babel (http://openbabel.org/) to convert them to the mol2 format. Subsequently, Avogadro was used to optimizing the geometry of the structures and saved to be docked into the binding pockets of GPER-1[39].
2.2 Binding Site Identification and Molecular Docking
Site Map was used to locate binding sites on the GPER-1 enzyme and rate them according to their potential as therapeutic targets[40]. Binding sites were cross validated using the MetaPocket 2.0[41], and Site Hound servers[42], which are preferred for site recognition due to their high prediction accuracy rate. G-1 and LNS8801 were docked individually into the binding pocket of GPER-1 using the UCSF Chimera software package[37]. The docking score calculations were generated using the Graphical User Interface of AutoDock Tools[43,44]. The docked conformations were computed using the Lamarckian Genetic method, commonly associated with trustworthiness and sufficiency[45,46]. The grid box had the following dimensions and coordinates: size x=10.0, y=15.874, z=10.0 with center x=-21.95, y=1.08, z=8.77. The binding scores and poses that exhibited the best-ranked conformation and the lowest binding free energies (-7.8kcal/mol for GPER-1-G-1 and -7.2kcal/mol for GPER-1- LNS8801) were chosen and saved for further study. The protein stability was assured in the MD simulations by adjusting the system’s protonation levels with Maestro Schrödinger[47-49], correcting essential hydrogen atoms, and capping neutral residues. The ligand-free system of GPER-1 was investigated utilizing validated simulation protocols, which have been widely employed in our previous research[50-53].
2.3 MD Simulations
2.3.1 Simulations Protocol
MD simulations were performed using the AMBER18 software and its Particle Mesh Ewald Molecular Dynamics module to investigate the conformational dynamics of unbound GPER-1 and its complexes with G-1 and its enantiomer LNS8801[54]. The FF14SB force-field was applied to parameterize the protein structures[55]. Prior to simulation, the GPER-1 protein was prepared using an in-house pdb4amber script, which facilitated necessary modifications, renaming, and protonation of histidine residues. The LEAP module was employed to generate accurate parameters and topology files for GPER-1, and system neutralization was carried out using the same tool. Subsequently, energy minimization was performed under a constraint potential of 500kcal/mol, involving a two-step process: partial minimization (2,500 steps) followed by full minimization (5,000 steps). Furthermore, a gradual temperature ramp-up from 0 to 300K was applied to the systems, allowing for thermal equilibration at 300K through an unconstrained simulation period of 1,000ps. The atmospheric pressure was maintained at 1bar using a Berendsen barostat[56]. To ensure further stability, a 500ns MD simulation was conducted to explore the structural consequences of interacting GPER-1 with G-1 and its enantiomer LNS8801[57].
2.3.2 Post-molecular Dynamics Simulation Analysis
The enzyme coordinates for unbound GPER-1 (Apo) as well as GPER-1 in complex with G-1 and LNS8801 were recorded at 1ps intervals during the MD simulations. The trajectories were subsequently analyzed using the integrated CPPTRAJ module in AMBER 18[58]. Post-analysis of the MD simulation systems involved evaluating various parameters including protein stability (RMSD), flexibility (RMSF), compactness (Rog), solvent accessible surface area (SASA), and dynamic cross-correlation (DCCM). Additionally, principal component analysis (PCA) was performed to elucidate the extent of atomic displacement in the enzyme. The binding free energies between the ligands and the protein were calculated using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) technique. The obtained data and resulting complexes were visualized using the Microcal Origin analytical tool. (https://www.originlab.com/)[59], NMW is implemented in Visual Molecular Dynamics (https://www.ks.uiuc.edu/Research/vmd/)[60].
2.4 MM/GBSA Ligands-protein Binding Free Energy Computations
The free energies of interaction, including polar and non-polar solvation energies, electrostatic interactions, and van der Waals forces, between GPER-1 and its ligands G-1 and LNS8801 were calculated using the Molecular Mechanics/Generalized-Born Surface Area (MM/GBSA) method[61-65]. The mathematical equation for MM/GBSA is:
$$ {\Delta }{{G}}_{{b}{i}{n}{d}}={{G}}_{{c}{o}{m}{p}{l}{e}{x}}–{{G}}_{{p}{r}{o}{t}{e}{i}{n}}–{{G}}_{{i}{n}{h}{i}{b}{i}{t}{o}{r}}\left(1\right)$$
$$ {\Delta }{{G}}_{{b}{i}{n}{d}}={{E}}_{{g}{a}{s}}+{{G}}_{{s}{o}{l}}–{T}{S}\left(2\right)$$
The Gbind is the summation of the gas and solvent energy minus entropy (TS).
$$ {{E}}_{{g}{a}{s}}={{E}}_{{i}{n}{t}}+{{E}}_{{v}{d}{w}}+{{E}}_{{e}{l}{e}}\left(3\right)$$
Where Egas denotes the sum of the AMBER force fields’ internal energy terms, including Eint for torsion, angle, and bond energies; Evdw for van der Waals energies associated with a covalent bond; and Eelec for electrostatic energies associated with a non-bonded pair.
The solvent energy calculation is represented by the following equation:
$$ {{G}}_{{s}{o}{l}}={{G}}_{{G}{B}}+{{G}}_{{S}{A}}\left(4\right)$$
$$ {{G}}_{{S}{A}}={\gamma }{S}{A}{S}{A}\left(5\right)$$
The polar (GGB) and non-polar (GSA) solvation effects were evaluated using the solvent-accessible surface area (SASA) approach. This involved employing a water probe with a radius of 1.4Å and setting the surface tension constants ‘b’ and ‘c’ to 0kcal/mol and 0.0072kcal/mol, respectively[66]. Additionally, to examine the stability of GPER-1, a per-residue energy decomposition analysis was conducted, enabling the determination of the potential energy contribution of individual residues within the catalytic site. This analysis provides valuable insights at the atomic level regarding the blocking efficacy of G-1 and LNS8801 and elucidates the mechanism by which these ligands interact with and target GPER-1.
2.5 DCCM
Dynamic cross-correlation analysis was applied to explore the dynamic fluctuations and motions in the backbone of GPER-1, specifically focusing on the α-carbon atoms. This analysis encompassed both the unbound state of GPER-1 and its complexes with G-1 and LNS8801[67]. The cross-correlation elements (i and j) for the Cα atoms were calculated using the following Equation 6:
$$ {{C}}_{{i}{j}}=\frac{<∆{r}{i}.∆{r}{j}>}{{({<∆{r}}_{{i}}^{2}><{∆{r}}_{{j}}^{2}>)}^{\frac{1}{2}}}\left(6\right)$$
where ri=Cαi= the MD trajectory’s constant time. Cij=1 represents highly correlated motions in the trajectory, and Cij=-1 represents highly anticorrelated motions. The motion discrepancy between 1 and -1 demonstrates that I and j motions are anti-correlated.
2.6 PCA
PCA, an established technique for investigating conformational dynamics in proteins, was utilized to characterize the residual motions of GPER-1 in its unbound state and when bound to G-1 and LNS8801[68]. Through dimensionality reduction, PCA identified correlated and anticorrelated fluctuations based on the molecular dynamics trajectories[69,70]. The eigenvectors, derived from the atomic coordinates, were employed to construct a positional covariance matrix (C) that facilitated the analysis of collective motions. The eigenvectors provided insight into the directionality of motion, while the corresponding eigenvalues conveyed information about the magnitude of these motions[71,72]. The positional covariance matrix C’s matrix elements were calculated using the following equation:
$$ {{C}}_{{i}}=⟨\left({{q}}_{{i}}-⟨{{q}}_{{i}}⟩\right)({{q}}_{{j}}-⟨{{q}}_{{j}}⟩)⟩\left({i},{j}={1,2},\dots ,3{N}\right)\left(7\right)$$
In this expression, N denotes the overall number of Cα atoms, qi and qj represent their corresponding cartesian coordinates, and I, j represent the i-th and j-th positions, respectively.
Following superimposing the MD trajectories onto a reference structure and performing a least-squares match, we estimated the average to remove the rotational and translational movements[73,74]. The eigenvectors and eigenvalues are calculated by transforming the symmetric matrix C into a diagonal matrix Λ of eigenvalues utilizing orthogonal coordinate transformation matrix T, as shown below.
$$ {Ʌ}={{T}}^{{T}}{{C}}_{{i}{j}}{T}\left(8\right)$$
here eigenvalues represent the total mean-square fluctuation of the system along each eigenvector, and eigenvectors indicate the directions of motion to (qi).
3 RESULTS AND DISCUSSION
3.1 MM/GBSA-based Interaction Analyses
The binding affinities of G-1 and LNS8801 towards GPER-1 were computed using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) technique. The therapeutic substance’s pharmacological (inhibitory) action is determined by its interaction within the pocket site of its biological target. The detailed molecular information provided by these estimated energies could be used to develop medications with higher binding affinity. The technique is affordable yet dynamic and provides the capacity to predict atomistic interactions between biological targets and their therapeutic molecules.
Table 1 displays the data, demonstrating that G-1 and LNS8801 bind favourably to GPER-1 with ∆G readings of -29.87kcal/mol, and -28.09kcal/mol, respectively. The likeness in the predicted ∆G scores could be indicative of binding mechanisms that are analogous in nature.
Table 1. MM/GBSA-based Binding Affinity Profile of G-1 and LNS8801 Bound to GPER-1
Systems |
Energy Components (kcal/mol) |
||||
∆Evdw |
∆Eele |
∆Ggas |
∆Gsol |
∆Gbind |
|
G-1 |
-39.52 0.05 |
-2.08 0.10 |
-41.60 0.11 |
11.73 0.09 |
-29.87 0.05 |
LNS8801 |
-37.71 0.08 |
-13.57 0.16 |
-51.29 0.20 |
23.20 0.14 |
-28.09 0.08 |
Notes: All energies are in kcal/mol. ∆Eele = electrostatic energy; ∆Evdw = van der Waals energy; ∆Gbind = total binding free energy; ∆Gsol = solvation free energy; ∆Ggas = gas phase free energy.
3.2 Decomposing Total Binding Free Energy of G-1 and LNS8801 when Bound to GPER-1 to Disentangle the Building Block Residues that Strengthen Binding Affinity
The evaluation of decomposed per-residue energy provides energy participation of individual residues that interacted with G-1, and LNS8801 at the binding sites of GPER-1 during the 500ns molecular dynamic simulation, shedding light on the crucial significance of some residues that contributed immensely to the interaction between G-1, and LNS8801 and GPER-1. Figure 2 shows that arginine contributed the most to the overall energy compared to the other amino acid residues.
As observed in Figures 2A, from GPER-1 and G-1 interaction Arg155, Arg253 and Arg254 in the GPER-1 binding exhibited the highest total energy contributions amongst other binding site amino acid residues equal to (-172.15kcal/mol), (-165.45kcal/mol), and (-174.62kcal/mol), respectively. Likewise, from GPER-1 and LNS8801 interaction Arg155, Arg251, Arg253 and Arg254 in GPER-1 binding site are residues with the highest total energy compared to other residues (-174.77kcal/mol), (-163.54kcal/mol), (-169.58kcal/mol), and (-172.46kcal/mol), respectively as demonstrated in Figures 2B. These results suggest that arginine is a key residue in driving the interaction between the GPER-1 and G-1, and LNS8801.
|
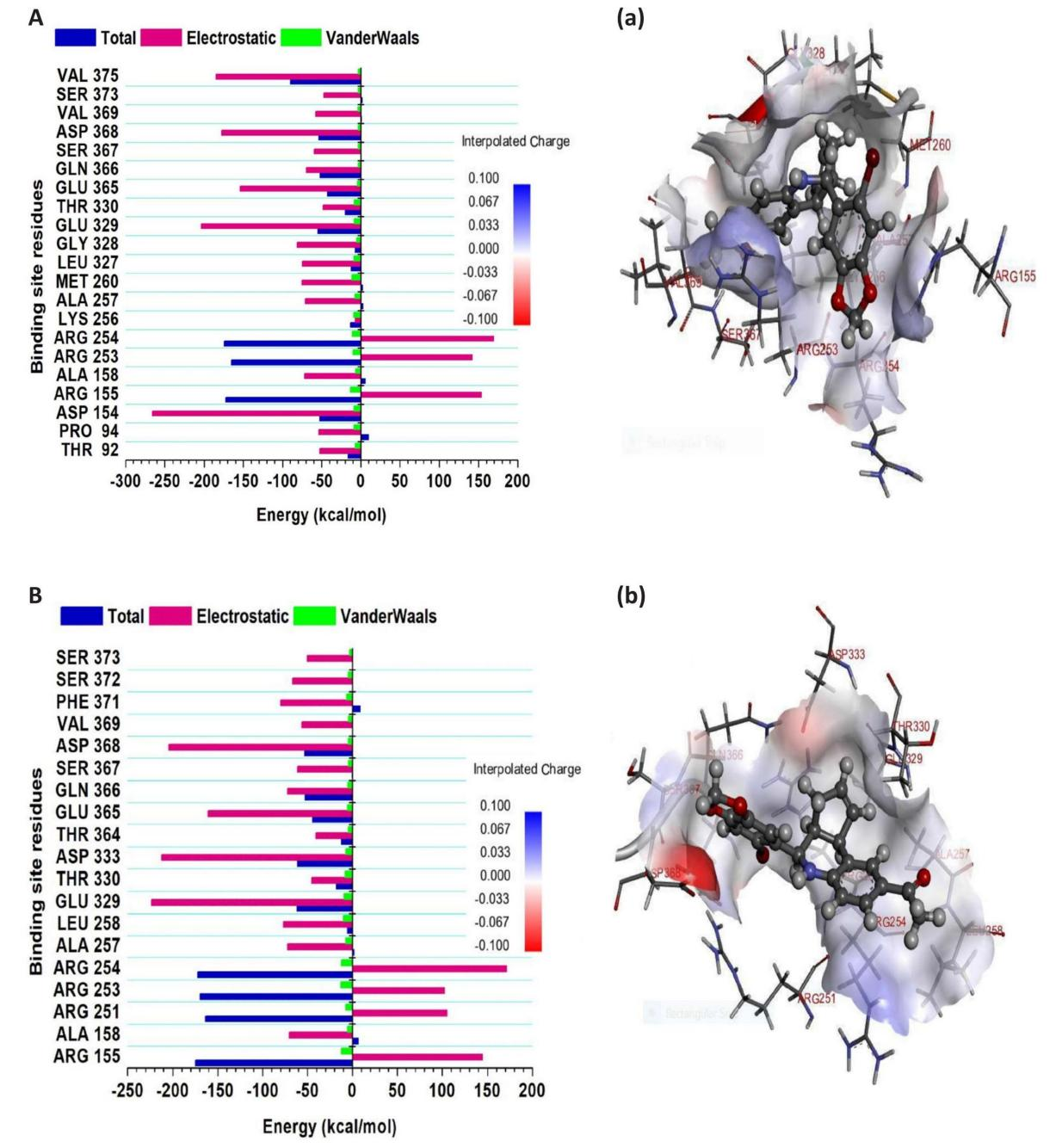
Figure 2. Plots illustrate Decomposed Per-residue Energy and their Participation in the Binding Affinity at the GPER-1’s Binding Pockets. A: when bound to G-1. B: when bound to LNS8801. Corresponding inter-molecular interactions are displayed in (a) G-1 (b) LNS8801.
3.3 Time-dependent Analysis of Important Interactions of G-1 and LNS8801 at the Binding Site of GPER-1
A time-dependent analysis of residues that established repeated interactions was performed to further investigate the interaction profile of G-1 and LNS8801 considering their stability inside the binding pocket of GPER-1. This analysis is significant as it provides valuable insights into the specific residues that play a crucial role in the binding and stability of G-1 and LNS8801 during the entire simulation period. Molecular interactions were visually examined at regular intervals during the 500ns molecular dynamics simulation. Representative snapshots were extracted at time points of 100, 200, 300, 400, and 500ns to ensure a comprehensive representation throughout the entire simulation duration. These selected snapshots offer a comprehensive depiction of the molecular interactions and dynamics observed during the simulation.
For the G-1 interactions with residues Arg253, Arg254 and Ala257 persisted throughout the entire 500ns simulation period indicating the key role these amino acid residues play in the binding and inhibitory process (Figure 3A). Amino acids Arg253 and Arg254 were among the top three performing residues exhibited the highest total energy contributions amongst other binding site amino acid residues (Figure 2A). Lys256 and Ser357 are residues that establish hydrogen bonds with carbonyl oxygen of acetophenone moiety of G-1, while Arg155 and Arg253 are residues that establish hydrogen bonds with one of the oxygens in dioxole moiety of G-1.
For the LNS8801 interactions with residues Arg253, Arg254 and Asp368 persisted throughout the entire 500ns simulation period indicating the key role these amino acid residues play in the binding and inhibitory process (Figure 3B). Amino acids Arg253 and Arg254 were among the top four performing residues exhibited the highest total energy contributions amongst other binding site amino acid residues (Figure 2B). Asp368 is the residue that establish hydrogen bond with one of the oxygens in dioxole moiety of LNS8801.
Collectively, findings here highlight the similarity in the binding patterns of the two inhibitors characterized by forming and sustaining interactions with key amino acids in the GPER-1 structure. The consistency of these residues in maintaining these interactions shows the active role they have towards GPER-1 inhibition. Interestingly, most of these implicated amino acid residues contributed the most energies towards the total free binding energy of all three inhibitors as reported earlier in the per residue energy contribution analysis. These findings highlight these residues as key drivers of GPER-1 inhibition.
|
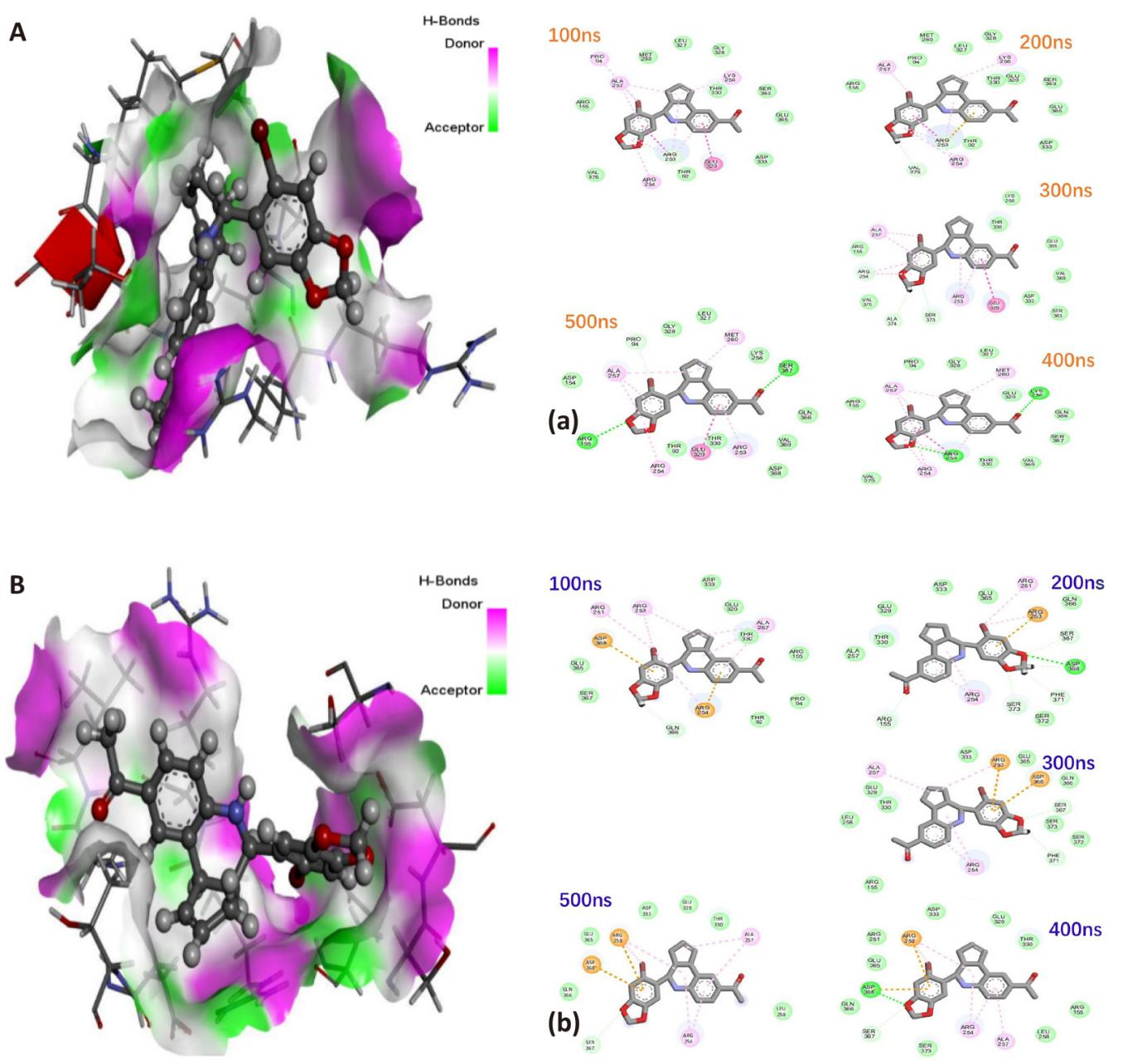
Figure 3. 3D Illustration of the Degree of Hydrogen Bond Donors (Magenta) and Acceptors (Light Green) Inside GPER-1 Active Site. A: When bound to G-1. B: When bound to LNS8801. Interaction dynamics represented in a comparative, time-based manner for GPER-1 coupled to (a) G-1, and (b) LNS8801.
3.4 Unveiling Conformational and Structural Changes in GPER-1 upon Binding of G-1 and LNS8801
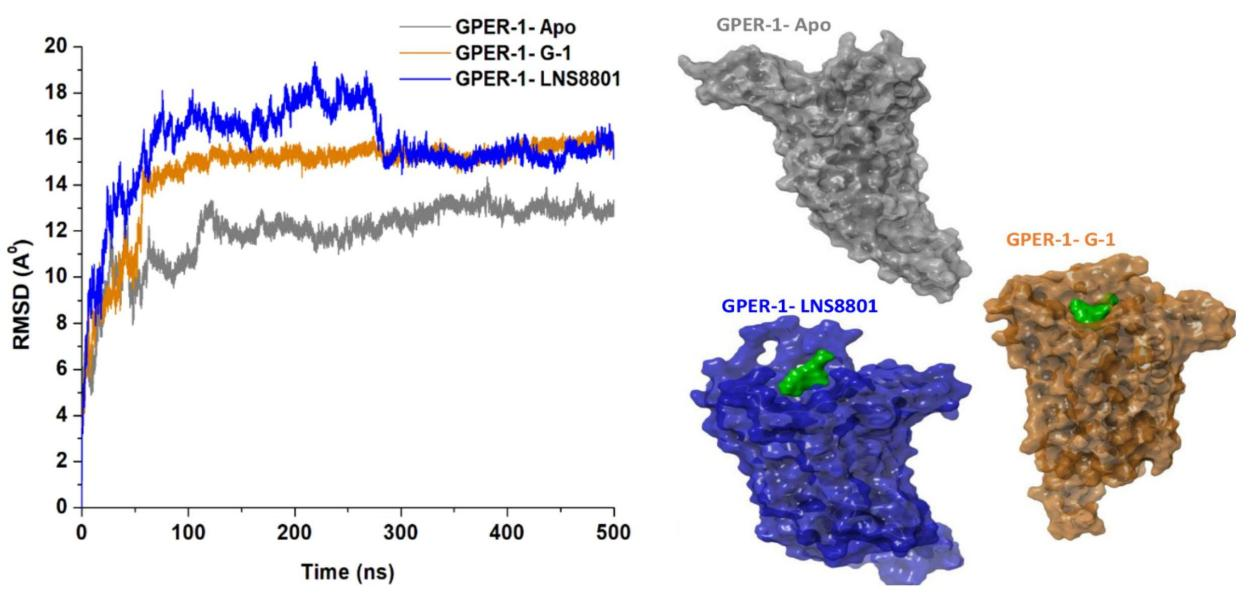
In order to ensure the reliability of the molecular dynamics (MD) simulations, the stability of the three systems was carefully evaluated to confirm the convergence and structural integrity of the ligand-bound and unbound states over the course of the 500ns simulation. Assessing the stability of atomic motions is crucial to identify and mitigate any undesired fluctuations or simulation defect[75]. To this end, the structural stability of each investigated system was quantified using the root-mean-square deviation (RMSD) of the C-α atoms. By comparing the protein conformation of GPER-1 in its bound states with G-1 and LNS8801 to its unbound form, the RMSD metric provided an estimate of the atomic deviations observed in the backbone structures of the protein backbone[76]. An elevated RMSD finding indicates that the backbone atoms are deviating more, whilst a low RMSD value usually indicates a reduction in the motions within the backbone atoms and could also afford an explanation for structural stability[52]. As revealed in Figure 4, the binding of G-1, and LNS8801 to the GPER-1 protein was seen to have consistently caused structural modification, presenting as increase in backbone atoms deviations in comparison to its apo form. The average RMSD for GPER-1, when bound to G-1, and LNS8801, were found to be 14.59Å, and 15.67Å. However, the median RMSD reading for peptide-free GPER-1 is 11.91Å. Consequently, we can infer that the binding of G-1, and LNS8801 to GPER-1 had activating impact since the bound systems demonstrated higher RMSD averages than the unbound system.
|
We further analysed the structural flexibility of GPER-1 protein as a consequence of the binding of G-1, and LNS8801. To investigate this, the C-α atoms root mean square fluctuation (RMSF) of GPER-1 when bound to these ligands compared to its ligand-free state was measured throughout the 500ns simulation period. An elevated RMSF worth generally indicates heightened fluctuations within the backbone structure of the protein, which ultimately leads to a more flexible structure and vice versa[77]. Figure 5 demonstrates that compared to the ligand-free state of GPER-1, the binding of G-1 decreased the GPER-1 fluctuation rates. However, the binding of LNS8801 increase fluctuations compared to the ligand-free system. The RMSF readings for the GPER-1 when bound to G-1, and LNS8801 were 3.39Å and 4.81Å respectively, while ligand-free GPER-1 had 3.55Å.
|
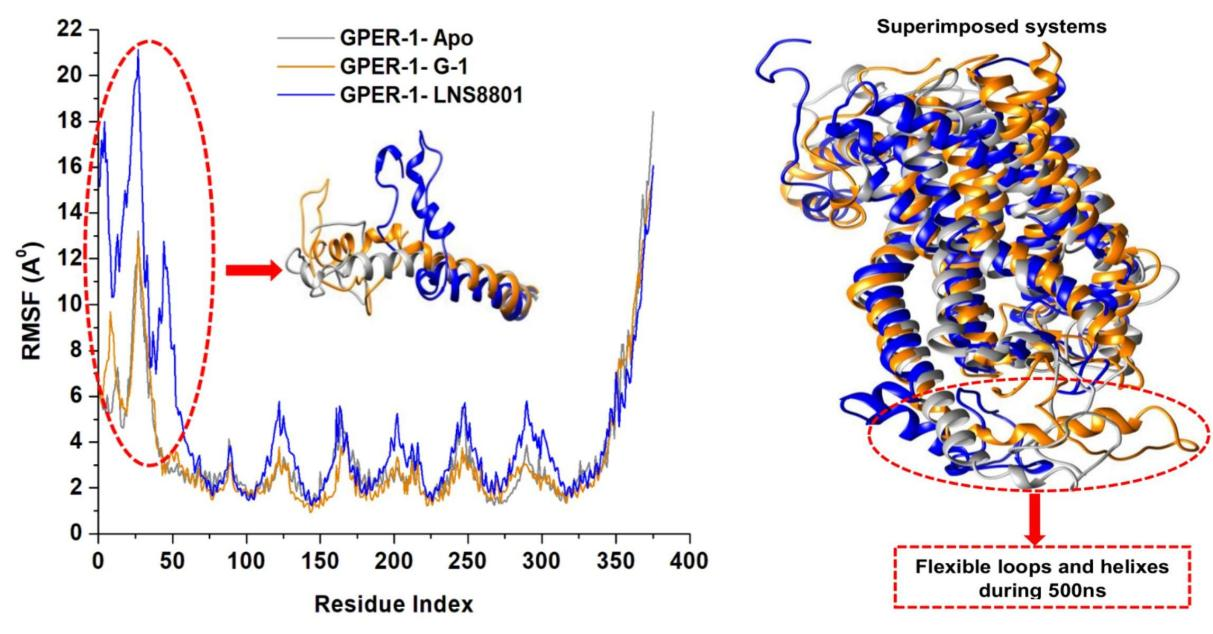
Figure 5. Time Evolution RMSF of Each Residue of the Protein C-α Atom over 500ns for Ligand-Free GPER-1 and When Bound to G-1 and LNS8801. Insert shows superimposed structures of the studied systems showing regions with high fluctuations (red circle).
Further to the RMSF computations, the radius of gyration (RoG) was explored to shed light on the compactness of GPER-1 when bound to G-1, and LNS compared to GPER-1 ligand-free state. RoG is determined by squaring the mass-weighted root-mean-square distance of the group of atoms distant from their shared center of mass[78,79]. The average Rg readings for the three systems, GPER-1-Apo (26.72Å), GPER-1- G-1 (25.52Å), and GPER-1-LNS8801 (25.46Å). These results demonstrated that the binding of G-1 and LNS8801 increase the rigidity of GPER-1 compared to ligand-free state (Figure 6).
. |
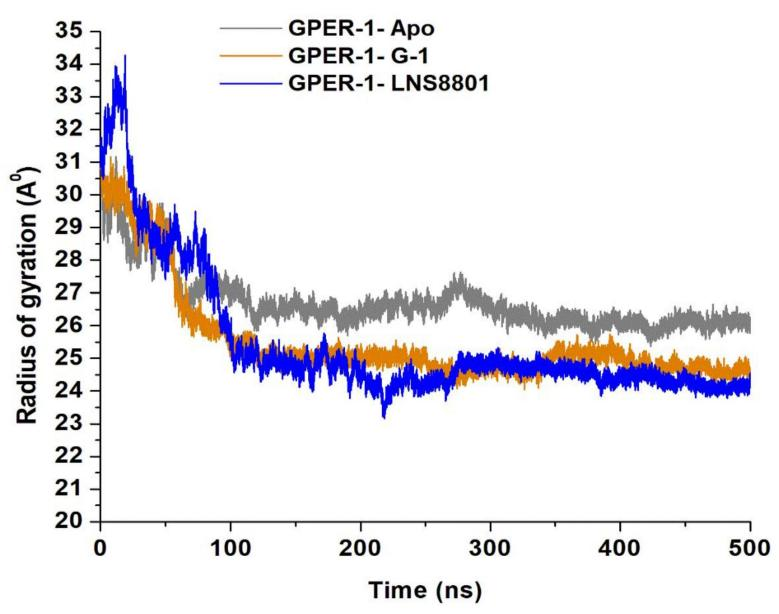
Figure 6. Radius of Gyration (ROG) of C-α Atoms of GPER-1-Apo, GPER-1-G-1, and GPER-1-LNS8801 Calculated from the MD Trajectories of 500ns.
Moreover, we proceeded to examine whether the exposure of residues in the presence of solvent molecules throughout the molecular dynamic simulation was affected by the binding of G-1, and LNS8801 and whether this exposure impacted specific interactions. Therefore, the solvent-accessible surface area (SASA) metrics were computed for each system[80]. As observed in Figure 7, the average SASA of GPER-1 in the ligand-free state is 22904.40Å2, while the average SASA of GPER-1 when bound to G-1, and LNS8801 are 22453.95Å2 and 22272.44Å2, respectively. The binding of G-1 and LNS8801 to GPER-1 induces a structural rearrangement of active site residues, which may explain the decrease in solvated area.
|
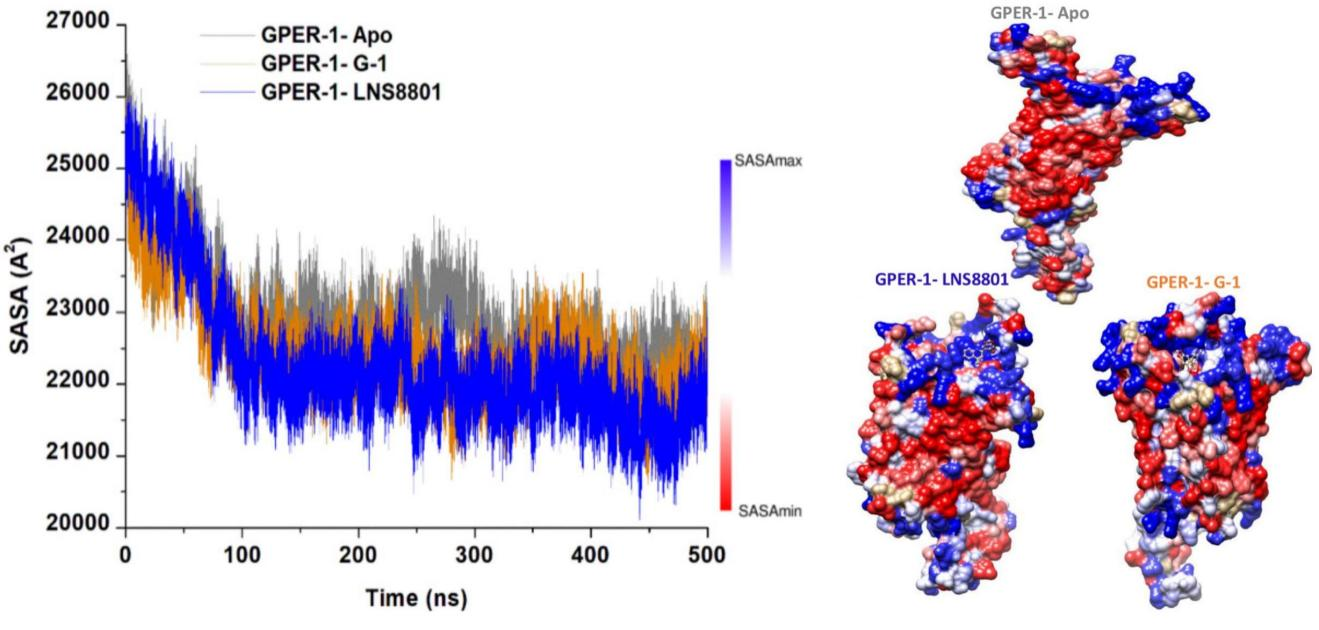
Figure 7. C-α Atoms SASA Relative to the Initial Minimized Structure Throughout 500ns for Ligand-Free GPER-1 and When Bound to G-1 and LNS8801. Red and blue indicate regions with lower and higher SASA values, respectively.
3.5 DCCM
In order to investigate the variations in the dynamics of GPER-1 in its Apo form and when bound to G-1 and LNS8801, dynamic cross-correlation matrices (DCCM) were constructed to analyze both anti-correlated and correlated protein structural motions (Figure 8). Scatter plots were utilized to depict the communities in which GPER-1 residues exhibited negative or positive correlations with respect to their residual motion. Throughout the simulation, both positive and negative movements were observed in the GPER-1-LNS8801 complex. In contrast, the GPER-1-Apo displayed a distinct pattern, characterized by a more pronounced positive correlation compared to the GPER-1-G-1 and GPER-1-LNS8801 complexes.
|
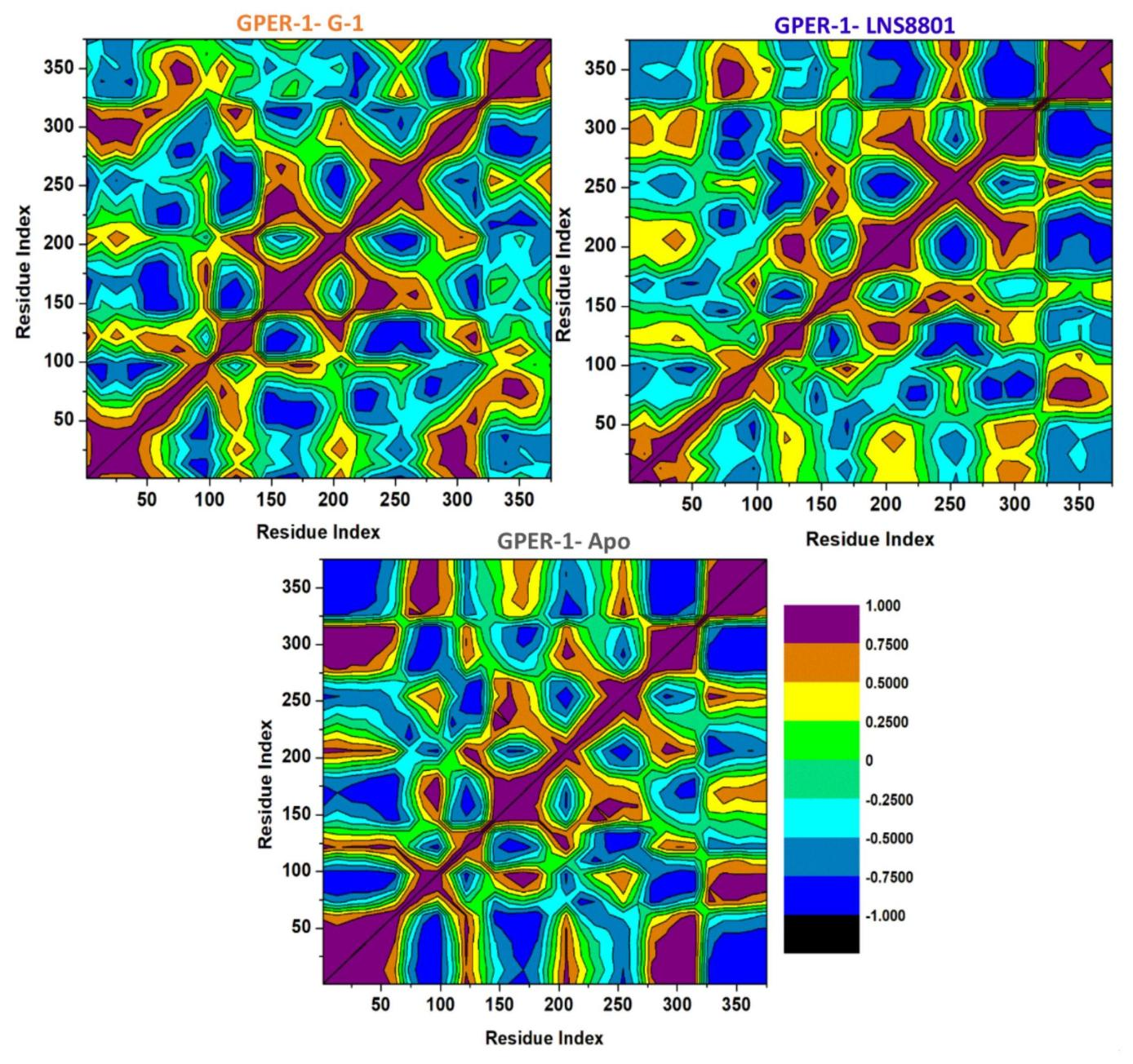
Figure 8. Dynamic Cross-Correlation Matrix Analyses for GPER-1 Ligand-Free State (Apo) and When Bound to G-1 and LNS8801 Calculated from the MD Trajectories of 500ns.
3.6 PCA
Intensive movements of GPER-1 when bound to G-1, and LNS8801 and when in free-peptide state were evaluated using PC analysis with the first two eigenvectors (EVs) to qualitatively examine the influence of induced mutations on the major conformational movements of each amino acid residue[53]. The conformational changes of bound and unbound CXCR4 were shown in a 2D scatter plot (Figure 9A), whereas the prominent PC1-using EVs in bound and unbound GPER-1 are depicted in (Figure 9B). Figure 9A shows 2D scatter plots for all three systems, indicating a significant change in GPER-1’s overall movements after being bounded to G-1, and LNS8801. Moreover, Figure 9A demonstrates that the GPER-1 when bound to G-1, imposed highly fluctuated anti-correlated effects as the negative values of 2D-scatter points into the protein. GPER-1 in a free state and, when bound to LNS8801, demonstrate prominent correlated motions with minimal system fluctuations. The scattered patterns in Figure 9B depict the transition of protein movements between states, with GPER-1-Apo demonstrating a higher transition than bound GPER-1.
|
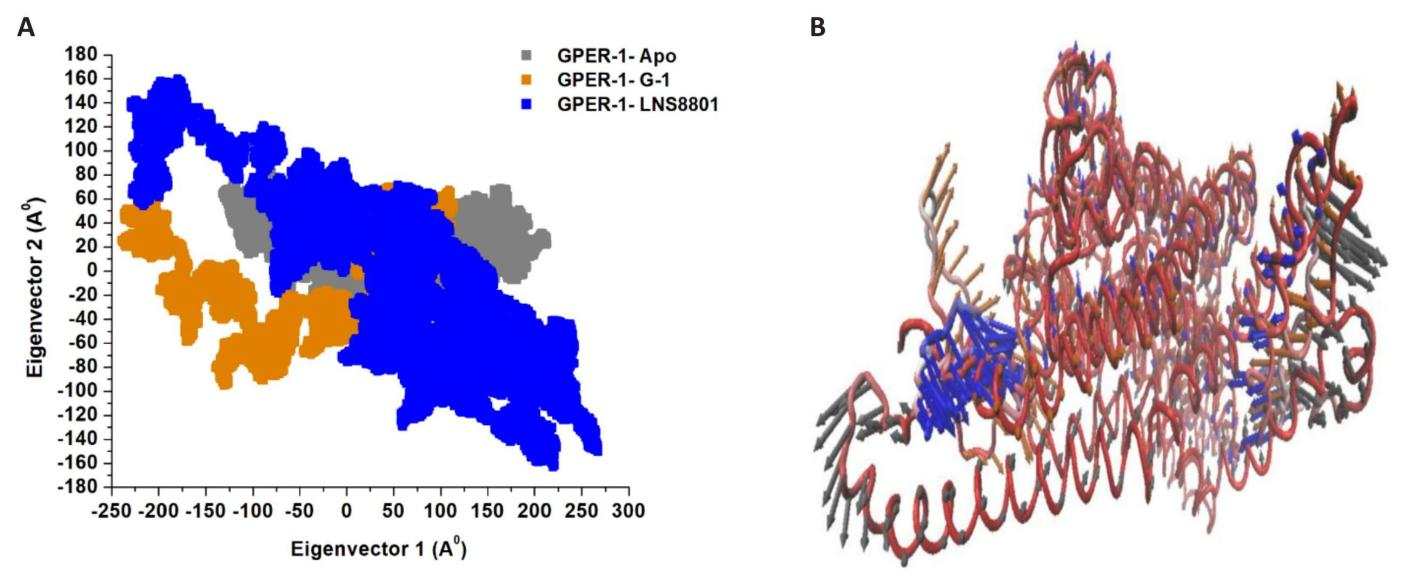
Figure 9. Principal Component Analysis. A: Apo GPER-1 and bound GPER-1 to G-1, and LNS8801. B: PC1 collective motions for the obtained predominant EVs utilizing PCA across the 500ns molecular dynamics simulation.
4 CONCLUSION
This study provides insight into the structural and conformational dynamics of GPER-1 upon binding of G-1 and its enantiomer LNS8801 and demonstrates that GPER-1 is a potential actionable target in WM. Based on the binding affinity, G-1, and LNS8801 demonstrated favourable binding to GPER-1 protein with ΔG values of -29.87kcal/mol, and -28.09kcal/mol, respectively. Furthermore, according to the per residue energy decomposition and time-dependent analysis, the amino acid residues Arg253 and Arg254 played a key role in the interactions between these ligands and GPER-1. They contributed significantly to the total binding affinity and consistently formed crucial interactions with the bound inhibitors throughout the entire 500ns simulation period. From a structural perspective, G-1, and LNS8801 affected the stability, flexibility, compactness, solvent-accessible surface area, and correlated motions of GPER-1. This was distinguished by several structural changes, both in the ligand-bound and ligand-free forms, that were able to impede GPER-1’s biological activity. Slight variations in the results, including binding affinity, indicate convergence in effectiveness with the superiority of G-1 over LNS8801. Overall, the study's findings shed light on important molecular information about the chirality of the G-1 and its enantiomer LNS8801 that could be applied to designing GPER-1 activators in the future. This allows scientists to pinpoint specific regions of a target protein that interact with the ligand of interest and address any structural obstacles that might prevent the desired effects from being realized. This helps scientists make informed predictions before spending time and money on laborious laboratory experiments.
Acknowledgements
The authors would like to acknowledge the Centre of High-Performance Computing (CHPC, www.chpc.ac.za), Cape Town, RSA, for the use of computational resources.
Conflicts of Interest
The authors declared no conflict of interest.
Data Availability
All data generated or analyzed during this study are included in this published article.
Copyright Permissions
Copyright © 2024 The Author(s). Published by Innovation Forever Publishing Group Limited. This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.
Author Contribution
Elamin G conceptualized, implemented, analyzed, interpreted and wrote the manuscript. Aljoundi A performed molecular dynamics simulation. Soliman MES set out project plan, concept, revised and approved the manuscript for submission.
Abbreviation List
AMBER, Assisted Model Building with Energy Refinement
CXCR4, CXC-motif-chemokine receptor 4
DCCM, Dynamic cross-correlation matrix
GPCR, G protein–coupled receptor
GPER-1, G protein-coupled estrogen receptor 1
MD, Molecular dynamics
MM/GBSA, Molecular mechanics/generalized Born surface area
PCA, Principal component analysis
RoG, Radius of gyration
RMSD, Root mean square deviation
RMSF, Root mean square fluctuation
SASA, Solvent accessible surface area
WM, Waldenström’s Macroglobulinemia
3D, Three-dimensional
References
[1] Kaiser LM, Hunter ZR, Treon SP et al. CXCR4 in Waldenström’s Macroglobulinema: chances and challenges. Leukemia, 2021; 35: 333–345.[DOI]
[2] Herrinton LJ, Weiss NS. Incidence of Waldenström’s macroglobulinemia. Blood, 1993; 82: 3148–3150.[DOI]
[3] Groves FD, Travis LB, Devesa SS et al. Waldenström’s macroglobulinemia: incidence patterns in the United States, 1988-1994. Cancer,1998; 82: 1078–1081.[DOI]
[4] Sekhar J, Sanfilippo K, Zhang Q et al. Waldenström macroglobulinemia: a Surveillance, Epidemiology, and End Results database review from 1988 to 2005. Leuk. Lymphoma, 2012; 53: 1625–1626.[DOI]
[5] Cingam S, Sidana S. Differential Diagnosis of Waldenström’s Macroglobulinemia and Early Management: Perspectives from Clinical Practice. Blood Lymphat Cancer Targets Ther, 2022; 2022: 107–117.[DOI]
[6] Treon S P, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. New Engl J Med, 2012; 367: 826–833.[DOI]
[7] Gopal AK, Kahl BS, De Vos S et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. New Engl J Med, 2014; 370: 1008–1018.[DOI]
[8] Treon SP, Tripsas CK, Meid K et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. New Engl J Med. 2015; 372: 1430–1440.[DOI]
[9] Kastritis E, Gavriatopoulou M, Kyrtsonis MC et al. Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenström macroglobulinemia: final analysis of a phase 2 study. Blood. 2015; 126: 1392–1394.[DOI]
[10] Treon SP, Gustine J, Meid K et al. Ibrutinib Monotherapy in Symptomatic, Treatment-Naïve Patients With Waldenström Macroglobulinemia. J Clin Oncol, 2018; 36: 2755–2761.[DOI]
[11] Roberts AW, Huang DCS. Targeting BCL2 With BH3 Mimetics: Basic Science and Clinical Application of Venetoclax in Chronic Lymphocytic Leukemia and Related B Cell Malignancies. Clin Pharmacol Ther. 2017; 101: 89–98.[DOI]
[12] Kaiser LM, Harms M, Sauter D et al. Targeting of CXCR4 by the Naturally Occurring CXCR4 Antagonist EPI-X4 in Waldenström’s Macroglobulinemia. Cancers (Basel), 2021; 13: 826.[DOI]
[13] O’Hayre M, Degese MS, Gutkind JS. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr Opin Cell Biol, 2014; 27: 126–135.[DOI]
[14] Hauser AS, Attwood MM, Rask-Andersen M er al. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov, 2017; 16: 829–842.[DOI]
[15] Treon SP, Hunter ZR. A new era for Waldenstrom macroglobulinemia: MYD88 L265P. Blood, 2013; 121: 4434–4436.[DOI]
[16] Li Y, Gong D, Zhang L et al. Zinc finger protein 32 promotes breast cancer stem cell-like properties through directly promoting GPER transcription. Cell Death Dis, 2018; 9:1162.[DOI]
[17] Lv X, He C, Huang C et al. G-1 Inhibits Breast Cancer Cell Growth via Targeting Colchicine-Binding Site of Tubulin to Interfere with Microtubule Assembly. Mol Cancer Ther, 2017; 16: 1080–1091.[DOI]
[18] Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol, 2011; 7: 715–726.[DOI]
[19] Madeo A, Maggiolini M. Nuclear Alternate Estrogen Receptor GPR30 Mediates 17β-Estradiol–Induced Gene Expression and Migration in Breast Cancer–Associated Fibroblasts. Cancer Res, 2010; 70: 6036–6046.[DOI]
[20] Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J. Endocrinol, 2010; 204: 105–114.[DOI]
[21] Natale CA, Li J, Zhang J et al. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. Elife, 2018; 7: e31770.[DOI]
[22] Harding AT, Goff MA, Froggatt HM et al. GPER1 is required to protect fetal health from maternal inflammation. Science, 2021;371: 271–276.[DOI]
[23] Morelli E, Hunter ZR, Fulciniti M et al. Therapeutic activation of G protein-coupled estrogen receptor 1 in Waldenström Macroglobulinemia. Exp Hematol Onco, 2022; 11: 1-6.[DOI]
[24] Maier NM, Franco P, Lindner W. Separation of enantiomers: needs, challenges, perspectives. J Chromatogr A, 2001; 906: 3–33.[DOI]
[25] Lorenz H, Seidel-Morgenstern A. Processes To Separate Enantiomers. Angew Chemie Int Ed, 2014; 53: 1218–1250.[DOI]
[26] Williams K, Lee E. Importance of Drug Enantiomers in Clinical Pharmacology. Drugs, 30: 333–354.[DOI]
[27] Rouhi AM. Chiral business. Chem Eng News Arc, 2003; 81: 45–61.[DOI]
[28] Coelho MM, Fernandes C, Remião F et al. Enantioselectivity in Drug Pharmacokinetics and Toxicity: Pharmacological Relevance and Analytical Methods. Molecules, 2021; 26: 3113.[DOI]
[29] Hosseinkhani H. Biomedical Engineering. Biomedical Engineering: Materials, Technology, and Applications. Wiley.2022.[DOI]
[30] Hosseinkhani H. Nanomaterials in Advanced Medicine. Wiley. 2019.
[31] Elamin G, Aljoundi A, Soliman MES. Co‐Binding of JQ1 and Venetoclax Exhibited Synergetic Inhibitory Effect for Cancer Therapy; Potential Line of Treatment for the Waldenström Macroglobulinemia Lymphoma. Chem Biodiver, 2022; 19: e202100845. [DOI]
[32] Usha T, Shanmugarajan D, Goyal AK et al. Recent Updates on Computer-aided Drug Discovery: Time for a Paradigm Shift. Curr Top Med Chem, 2018; 17: 3296–3307.[DOI]
[33] Varadi M, Anyango S, Deshpande M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Re, 2022; 50: D439–D444.[DOI]
[34] Jumper J, Evans R, Pritzel A et al. Highly accurate protein structure prediction with AlphaFold. Nature, 2021; 596: 583–589.[DOI]
[35] Bateman A, Martin MJ, O’Donovan C et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res, 2017; 45: D158–D169.[DOI]
[36] Kusumaningrum S, Budianto E, Kosela S et al. The molecular docking of 1,4-naphthoquinone derivatives as inhibitors of Polo-like kinase 1 using Molegro Virtual Docker. J Appl Pharm Sci, 2014; 4: 047-053.
[37] Pettersen EF, Goddard TD, Huang CC et al. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem, 2004; 25: 1605–1612.[DOI]
[38] Kim S, Thiessen PA, Bolton EE et al. PubChem Substance and Compound databases. Nucleic Acids Res, 2016; 44: D1202–D1213.[DOI]
[39] Hanwell MD, Curtis DE, Lonie DC et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J Cheminform, 2012; 4: 1–17.[DOI]
[40] Halgren TA. Identifying and Characterizing Binding Sites and Assessing Druggability. J Chem Inf Model, 2009; 49: 377–389.[DOI]
[41] Huang B. MetaPocket: A Meta Approach to Improve Protein Ligand Binding Site Prediction. Omi. A J. Integr. Biol, 2009; 13: 325–330.[DOI]
[42] Hernandez M, Ghersi D, Sanchez R. SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res, 2009; 37(suppl_2): W413–W416.[DOI]
[43] Allouche A-R. Gabedit-A graphical user interface for computational chemistry softwares. J Comput Chem, 2011; 32: 174–182.[DOI]
[44] Aljoundi A, Bjij I, El Rashedy A et al. Covalent Versus Non-covalent Enzyme Inhibition: Which Route Should We Take? A Justification of the Good and Bad from Molecular Modelling Perspective. Protein J, 2020; 39: 97–105.[DOI]
[45] Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem, 2010: 31: 455–461.[DOI]
[46] Aljoundi AK, Agoni C, Olotu FA et al. Turning to Computer-aided Drug Design in the Treatment of Diffuse Large B-cell Lymphoma: Has it been Helpful? Anticancer. Agents Med Chem, 2019; 19: 1325–1339.[DOI]
[47] Madhavi Sastry G, Adzhigirey M, Day T et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des, 2013; 27: 221–234.[DOI]
[48] Aljoundi A, El Rashedy A, Appiah-Kubi P et al. Coupling of HSP72 α-Helix Subdomains by the Unexpected Irreversible Targeting of Lysine-56 over Cysteine-17; Coevolution of Covalent Bonding. Molecules, 2020; 25: 4239.[DOI]
[49] Mohamed AI, Beseni BK, Msomi NZ et al. The antioxidant and antidiabetic potentials of polyphenolic-rich extracts of Cyperus rotundus (Linn.). J Biomol Struct Dyn, 2022 40: 12075–12087.[DOI]
[50] Elamin G, Aljoundi A, Soliman MES. A synergistic multitargeted of BET and HDAC: an intra-molecular mechanism of communication in treatment of Waldenström macroglobulinemia. Mol Simul, 2022; 48: 197–208.[DOI]
[51] Elamin G, Aljoundi A, Soliman MES. Multi-catalytic Sites Inhibition of Bcl2 Induces Expanding of Hydrophobic Groove: A New Avenue Towards Waldenström Macroglobulinemia Therapy. Protein J, 2022; 41:201–205.[DOI]
[52] Elamin G, Aljoundi A, Alahmdi MI et al. Battling BTK mutants with noncovalent inhibitors that overcome Cys481 and Thr474 mutations in Waldenström macroglobulinemia therapy: structural mechanistic insights on the role of fenebrutinib. J Mol Model, 2022; 28: 355.[DOI]
[53] Elamin G, Aljoundi A, Alahmdi M et al. Revealing the Role of the Arg and Lys in Shifting Paradigm from BTK Selective inhibition to the BTK/HCK Dual inhibition- Delving into the Inhibitory Activity of KIN-8194 Against BTK, and HCK in the Treatment of Mutated [BTK]^ Cys481 Waldenström Macroglobul. Anticancer Agents Med Chem, 2023.[DOI]
[54] Case DA, Cheatham TE, Darden T et al. The Amber biomolecular simulation programs. J Comput Chem, 2005; 26: 1668–1688.[DOI]
[55] Wang J, Wolf RM, Caldwell JW et al. Development and testing of a general amber force field. J Comput Chem, 2004; 25: 1157–1174.[DOI]
[56] Berendsen HJC, Postma JPM, van Gunsteren WF et al. Molecular dynamics with coupling to an external bath. J Chem Phys, 1984; 81: 3684–3690.[DOI]
[57] Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys, 1977; 23: 327–341.[DOI]
[58] Roe DR, PTRAJ TEC. CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Compu, 2013; 9: 3084–3095.[DOI]
[59] Seifert E. OriginPro 9.1: scientific data analysis and graphing software-software review. J Chem Inf Model, 2014; 54: 1552.[DOI]
[60] Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph, 1996; 14: 33–38.[DOI]
[61] Hou T, Wang J, Li Y et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model, 2011; 51: 69–82.[DOI]
[62] Homeyer N, Gohlke H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol Inform, 2012; 31: 114–122.[DOI]
[63] Genheden S, Kuhn O, Mikulskis P et al. The Normal-Mode Entropy in the MM/GBSA Method: Effect of System Truncation, Buffer Region, and Dielectric Constant. J Chem Inf Model, 2012; 52: 2079–2088.[DOI]
[64] Ylilauri M, Pentikäinen OT. MMGBSA As a Tool To Understand the Binding Affinities of Filamin–Peptide Interactions. J Chem Inf Model, 2013; 53: 2626–2633.[DOI]
[65] Massova I, Kollman PA. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect Drug Discov Des, 2000; 18: 113–135.[DOI]
[66] Sitkoff D, Sharp KA, Honig B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J Phys Chem, 1994; 98: 1978–1988.[DOI]
[67] Kasahara K, Fukuda I, Nakamura H. A Novel Approach of Dynamic Cross Correlation Analysis on Molecular Dynamics Simulations and Its Application to Ets1 Dimer–DNA Complex. PLoS One, 2014; 9: e112419.[DOI]
[68] Wan H, Hu JP, Tian XH et al. Molecular dynamics simulations of wild type and mutants of human complement receptor 2 complexed with C3d. Phys Chem Chem Phys, 2013; 15: 1241–1251.[DOI]
[69] Chang S, Hu JP, Lin PY et al. Substrate recognition and transport behavior analyses of amino acid antiporter with coarse-grained models. Mol Biosyst, 2010; 6: 2430–2438.[DOI]
[70] Fakhar Z, Govender T, Maguire GEMM et al. Differential flap dynamics in transpeptidase2 from mycobacterium tuberculosis revealed by molecular dynamics. Mol Biosyst, 2014; 13: 1223–1234.[DOI]
[71] David CC, Jacobs DJ. Principal Component Analysis: A Method for Determining the Essential Dynamics of Proteins. In: Livesay D (eds). Protein Dynamics. Methods in Molecular Biology (MIMB, volume 1084). Humana Press, Totowa, NJ, USA. 2014: 193–226.[DOI]
[72] Levy RM, Srinivasan AR, Olson WK et al. Quasi-harmonic method for studying very low frequency modes in proteins. Biopolymers, 1984; 23: 1099–1112.[DOI]
[73] Chen J, Wang J, Zhu W. Binding Modes of Three Inhibitors 8CA, F8A and I4A to A-FABP Studied Based on Molecular Dynamics Simulation. PLoS One, 2014; 9: e99862.[DOI]
[74] Ichiye T, Karplus M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins Struct Funct Genet, 1991; 11: 205–217.[DOI]
[75] Brüschweiler R. Efficient RMSD measures for the comparison of two molecular ensembles. Root-mean-square deviation. Proteins, 2003; 50: 26–34. [DOI]
[76] Pitera JW. Expected Distributions of Root-Mean-Square Positional Deviations in Proteins. J Phys Chem B, 2014; 118: 6526–6530.[DOI]
[77] Teilum K, Olsen JG, Kragelund BB. Functional aspects of protein flexibility. Cell Mol Life Sci, 2009; 66: 2231–2247.[DOI]
[78] Lobanov MY, Bogatyreva NS, Galzitskaya OV. Radius of gyration as an indicator of protein structure compactness. Mol Biol, 2008; 42: 623–628.[DOI]
[79] Salleh AB, Rahim A, Rahman R et al. The Role of Arg157Ser in Improving the Compactness and Stability of ARM Lipase. J Comput Sci Syst Biol, 2012; 5: 38–46.[DOI]
[80] Gromiha M, Ahmad S. Role of Solvent Accessibility in Structure Based Drug Design. Curr Comput Aided-Drug Des, 2005; 1: 223–235.[DOI]
Brief of Corresponding Author(s)
Mahmoud E.S. Soliman He is a Full Professor at the Department of Pharmaceutical Sciences, School of Health Sciences, UKZN, Westville Campus, and the Head and Principal Investigator of Molecular Bio-Computation and Drug Design Laboratory. He completed his postgraduate degree (MPhil/PhD - 2009) at the University of Bath, United Kingdom. Prof. Soliman’s research mainly focuses on studying biomolecular systems, drug-protein interactions, and rational design of drug candidates using experimental and computational techniques. He has published more than 320 publications in internationally accredited journals. Soliman accumulated H-index of 37. |



