Integrative Analysis Identified rs907091 at the miRNA Binding Region of the IKZF3 Gene as a Potential Functional Variant for Rheumatoid Arthritis
Wei Xia1,2,3, Longfei Wu1,3, Shufeng Lei1,3*
1Center for Genetic Epidemiology and Genomics, School of Public Health, Medical College of Soochow University, Suzhou, Jiangsu Province, China
2Zhenjiang Health Inspection Bureau, Zhenjiang, Jiangsu Province, China
3Jiangsu Key Laboratory of Preventive and Translational Medicine for Geriatric Diseases, Soochow University, Suzhou, Jiangsu Province, China
*Correspondence to: Shufeng Lei, PhD, Professor, Director, Center for Genetic Epidemiology and Genomics, School of Public Health, Medical College of Soochow University, Suzhou, 215123, Jiangsu Province, China; Email: leisf@suda.edu.cn
DOI: 10.53964/cme.2024007
Abstract
Objective: Previous studies have identified a large number of associations between genetic variants and rheumatoid arthritis (RA), but the functional mechanisms underlying the associations are largely unknown.
Methods: Based on publicly available datasets and results, we explored the functional mechanisms underlying the associations between regulatory polymorphisms in miRNA target sites (poly-miRTSs) and RA by combining integrative analyses (expression quantitative trait locus analysis, eQTL, and gene enrichment analysis) with in-house molecular experiments (allelic expression imbalance experiment, AEI, and dual-luciferase reporter gene assay).
Results: By integrating the results from the MirSNP and eQTL databases, a total of 114 pairs between poly-miRTSs and target genes were identified, which correspond to 70 unique poly-miRTSs and 28 genes. Most of the 28 genes were located in the HLA region, and they tended to be enriched in multiple immune-related GO terms or pathways, e.g., “antigen processing and presentation” and “immune response”. Among these poly-miRTSs, rs907091 in the 3’UTR of IKZF3 (non-HLA region) was highlighted. Further AEI experiments showed that the C allele had a higher expression of ~18% than the T allele, suggesting an obvious allelic expression imbalance. Dual-luciferase reporter gene assays showed that miR-326 and miR-330-5p could regulate IKZF3 expression (P<0.05) mediated by rs907091 in the 3’UTR binding site.
Conclusion: Our study explored some functional mechanisms underlying the associations between poly-miRTSs and target genes and highlighted rs907091 at the miRNA binding region of the IKZF3 gene as a potential functional variant for rheumatoid arthritis.
Keywords: rs907091, IKZF3, rheumatoid arthritis, miRNA, eQTL
1 INTRODUCTION
Rheumatoid arthritis (RA) is a complex autoimmune disease characterized by systemic inflammation affecting multiple peripheral joints that can progress to destruction of the cartilage and bone, progressive deformity, and severe disability. It is a challenge to identify specific RA genetic factors due to the complex nature of the genetic pathogenic mechanism. To date, great attempts have been made to identify genetic susceptibility to RA[1-6] through genome-wide association studies (GWASs)[2,4,7,8] and meta-analysis studies[9-12]. However, most of those studies only set up the statistical associations between genetic markers and RA at the DNA level without exploring the underlying functional mechanisms. Such established associations usually do not provide further insights into the functions of genetic markers or the regulation of gene expression that can link genetic information to the RA phenotype directly.
MicroRNAs (miRNAs) are endogenous noncoding regulatory RNAs containing 21 to 23 nucleotides that can bind to the 3’-untranslated regions (3’-UTRs) of target mRNAs to regulate posttranscriptional gene expression[13,14]. Therefore, polymorphisms in miRNA binding sites of 3’-UTRs could potentially influence the affinity between miRNAs and their target mRNAs[15,16]. This could result in allelic-specific regulation efficiency of miRNAs by altering their capacity to repress mRNA translation or promote mRNA decay[15,17]. Recently, many studies have shown that regulatory polymorphisms in miRNA target sites (poly-miRTSs) may play vital roles in a wide range of biological and disease pathogenesis[18-22].
Currently, numerous publicly available databases, e.g., GWAS meta-analysis[23], expression quantitative trait locus (eQTL) datasets[24-26], and poly-miRTS databases[27-29], can serve as important supplementary resources for further data mining to provide additional functional evidence to explain the functional mechanisms underlying the associations.
Based on previous results and available public databases, our study aimed to explore the functional mechanisms underlying the associations between the selected poly-miRTSs and RA by combining integrative analyses (eQTL analysis, and gene enrichment analysis) with in-house molecular experiments (allelic expression imbalance, AEI, and dual-luciferase reporter gene assay).
2 MATERIALS AND METHODS
2.1 Identification of Genetic Variants in miRNA-binding Sites
We first downloaded the association results between genetic variants and RA from recent large GWAS meta-analyses that comprehensively combined the results from 22 RA GWAS[23]. More than 8 million single nucleotide polymorphisms (SNPs) in Europe and more than 6 million SNPs in Asia were investigated in this study. We first selected significant SNPs with raw P values (P<5E-8) in Asia for the next step. We chose a relatively loose threshold because we tried to avoid missing potential loci at the beginning of our study. In this study, we only focused on the regulatory genetic variants in miRNA target sites.
The MirSNP (http://bioinfo.bjmu.edu.cn/mirsnp/search/) is a database of archiving over 410,000 human SNPs in predicted miRNA-mRNA binding sites[27]. In this database, annotations about whether a SNP within the target site would decrease/break or enhance/create a miRNA-mRNA binding site were available[27]. Therefore, the SNPs collected in the MirSNP probably contribute to the specific expression of target mRNA by binding miRNA. According to the SNP ID, we searched the above selected SNPs from GWAS meta-analyses in the MirSNP and identified the overlapping SNPs for the following eQTL analysis.
2.2 eQTL Analysis
eQTL analysis is a commonly used and powerful method of testing whether the identified functional SNPs may lead to variations in the mRNA expression of nearby genes. Recently, an increasing number of studies have identified a large number of putative eQTLs in multiple RA-related cells or tissues, including T cells, monocytes, lymphoblastoid cell lines, and fibroblasts[30], which were summarized and archived in an online available database for convenient and quick searching and comparison (eQTL: http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/). According to the above identified poly-miRTSs and corresponding genes, we searched the database and identified the significant pairs based on the significant eQTL results.
2.3 GO and KEGG Gene Enrichment Analysis
To further analyze the biological functions of the identified corresponding genes and assist in selecting potential loci for subsequent molecular experiments, we performed GO and KEGG gene enrichment analysis using the Database for Annotation, Visualization and Integrated Discovery integrated database query tools (http://david.abcc.ncifcrf.gov/)[31]. Furthermore, we also conducted protein–protein interaction (PPI) analysis in the Search Tool for the Retrieval of Interacting Genes (STRING) database (https://string-db.org/) to explore the potential interactions of identified genes[32]. In our study, the STRING 10.5 online tool was used to construct the PPI networks with the required confidence (combined score)>0.4.
2.4 Allelic Expression Imbalance Experiment
To explore the functional links for the identified pairs in-depth, we performed the following in-house molecular experiments (AEI and dual-luciferase reporter gene assay). Due to limited funds, we selected only one pair by considering the following factors: the association P value in GWAS studies, 3’ UTR of miRNA binding sites, eQTL results, SNP location at non-HLA region, and known gene functions associated with immune diseases. The corresponding miRNA target genes were simultaneously predicted by multiple online miRNA prediction tools and target prediction algorithms, including TargetScan (http://TargetScan.org/), miRanda (www.mirtoolsgallery.org/miRToolsGallery/node/1055), PITA (https://genie.weizmann.ac.il/pubs/mir07/mir07_data.html) and miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/).
AEI experiments are a powerful technique for identifying cis-regulatory SNPs controlling gene expression between two allelic transcripts. To verify whether the functional SNP can regulate its target gene expression, AEI experiments were performed in the mixed sample consisting of 1 genomic DNA and 2 cDNAs that have different alleles from the peripheral blood of one subject who had a heterozygous genotype of the target SNP. The forward and reverse primers were designed using the online software Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/). Genomic DNA from peripheral blood cells was isolated by using a Puregene DNA extraction kit (Qiagen, Valencia, CA), and total RNA was extracted with an RNeasy Mini Kit (Qiagen, Valencia, CA). Target mRNA was reverse transcribed using a Qiagen QuantiTect RT kit (Qiagen, Valencia, CA) following the manufacturer’s protocol, which can remove genomic DNA (gDNA) contamination by a DNase digestion step. Genotyping of DNA and cDNA was performed using GeneMapper 4.1 (Applied Biosystems Co., Ltd., USA), (the used primers: rs907091F: 5’-AGGCTTTTCCACGTGTGGTTGA-3’; rs907091R: 5’-TGTGAAACCTTTGTTTTCCCATCA-3’; rs907091cR: 5’-CAATGCCTCAGAAATTCATTGGTG-3’) and the single nucleotide extension method was used to quantify allele-specific cDNA and DNA using the ABI PRISM SNaPshot Multiplex Kit (Applied Biosystems Co., Ltd., USA) according to the manufacturer’s instructions (the used primer: rs907091SR: 5’-TTTTTTTTTTTTTTGCAGTGGAATGAGTGGTCCC-3’). Fluorescent PCR followed by capillary electrophoresis was used to quantify the SNP allele-specific transcripts of the target gene. The ratio of cDNA quantization between the two alleles was calculated, followed by normalization to the ratio of DNA quantization from the same subject.
2.5 Dual-Luciferase Reporter Gene Assays
Furthermore, we used dual-luciferase reporter gene assays to detect whether the rs907091 SNP located in the 3’-UTR of the IKZF3 gene alters the expression of IKZF3 mediated by miRNAs. First, we cloned a 601 bp region around the SNP from the 3’-UTR of the IKZF3 gene, and the insert fragment representing the T and C alleles of rs907091 was generated by PCR amplification of genomic DNA from two individuals homozygous for each allele. Then, we connected the target fragments into the dual-luciferase reporter plasmid vector PmirGLO (Promega, Wisconsin, USA). Finally, the ligated vectors were transformed into DH5α Escherichia coli-competent cells after growth at 37°C for 16 hours, and positive clones were selected by antibiotic resistance and further confirmed by PCR amplification and restriction digestion of plasmids. The construct sequences of the plasmids were verified by DNA sequencing before transfection. 293T cells were cultivated in antibiotic-free DMEM supplemented with 10% FBS in 96-well compatible plates at 37°C and 5% CO2 for transient transfection reporter assays. LipofectamineTM 2000 Transfection Reagent (Invitrogen, CA, USA) was used to transfect the plasmid and miRNAs into 293T cells according to the manufacturer’s instructions. Luciferase values were normalized to the Renilla luciferase value to adjust the transfection efficiencies. All experiments were repeated three times, and each experiment included five biological replicates for each allele.
3 RESULTS
According to the downloaded GWAS association results from Asia, we selected 11,186 significant SNPs (P<5e-8) for further functional annotations. Then, according to the SNP ID, we searched the MirSNP database and finally identified 253 overlapping SNPs that were used in the next eQTL analysis. According to the SNP ID, we searched the eQTL database, and a total of 114 pairs between SNPs and corresponding genes were identified, which correspond to 70 unique SNPs and 28 genes (Table S1). As we expected, most of the 28 genes are located in the HLA region, which plays an important role in the pathogenesis of many autoimmune diseases, including RA. The majority of RA-associated significant eQTLs were identified in RA-related cells (monocytes and lymphoblastoid cells).
Table S2 listed the top 15 significant GO terms. The 28 identified genes were enriched in multiple immune-related GO terms, including “integral component of luminal side of endoplasmic reticulum membrane”, “MHC class II protein complex”, “antigen processing and presentation”, and “immune response”. In addition, the identified genes were enriched in the pathways of multiple autoimmune diseases (Table S3), e.g., “Type I diabetes mellitus”, “Autoimmune thyroid disease”, “Inflammatory bowel disease” and “Rheumatoid arthritis”. Figure 1 shows the results of PPI analysis for the 28 identified genes. The large complex network consisted of multiple interacting HLA region genes. Another small network contains three genes (IKZF3, GSDMB and ORMDL3).
|
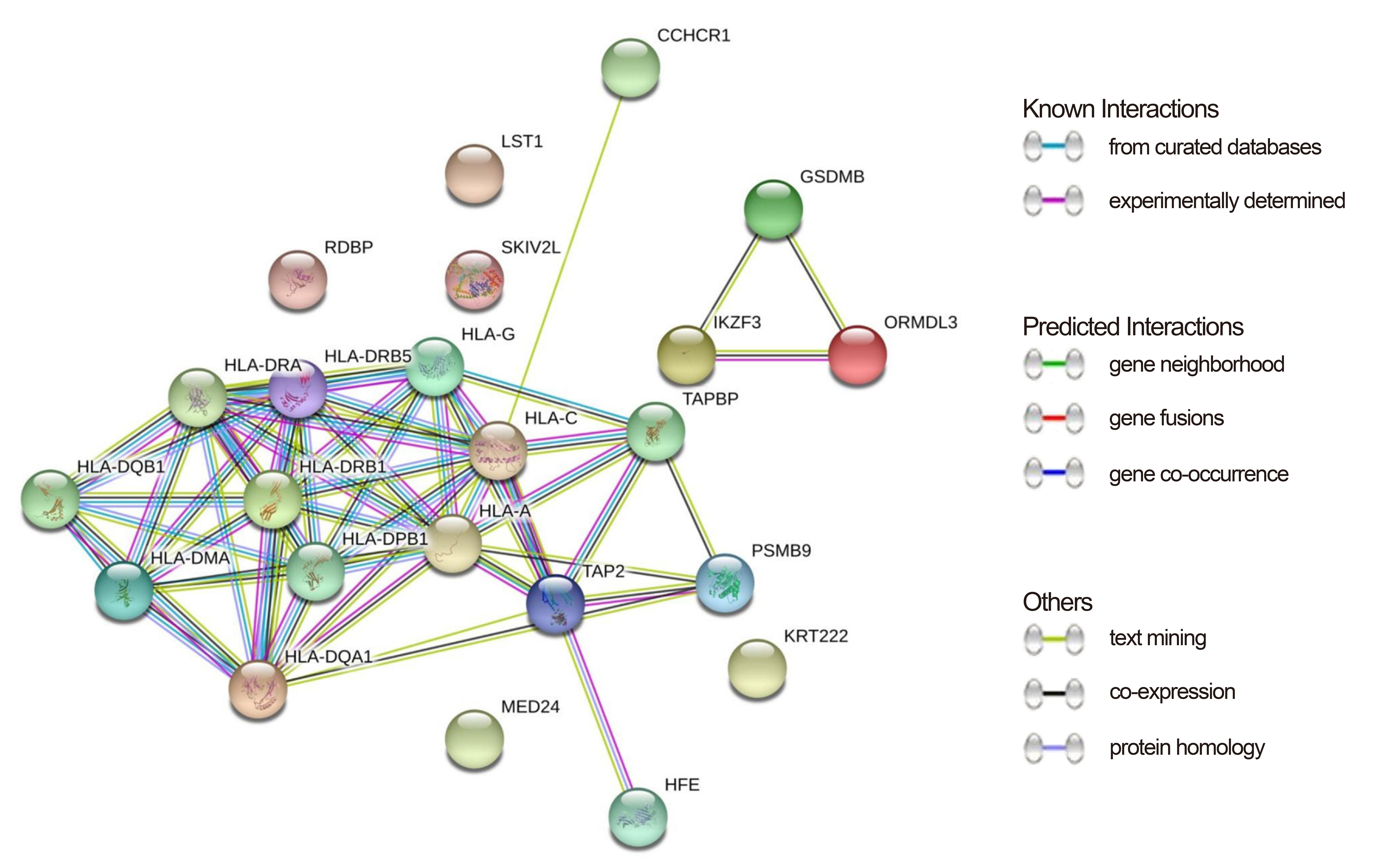
Figure 1. PPI analysis for the 28 identified genes from eQTL analysis. According to the SNP ID, we searched the MirSNP database and finally identified 253 overlapping SNPs. Furthermore, according to the SNP ID, we searched the eQTL database, and a total of 114 pairs between SNPs and corresponding genes were identified, which correspond to 70 unique SNPs and 28 genes. PPI analysis was performed according to the 28 identified genes. The colorful lines between genes indicate different connection types.
Furthermore, according to the regulatory polymorphisms in miRNA target sites (poly-miRTSs), we identified 32 pairs in which the SNPs were located in the 3’UTR of their eQTL genes among the above 114 pairs, which corresponded to 32 unique SNPs and 5 genes (Table 1). As shown in Table 1, HLA-DPB1 was the most frequently observed target eQTL gene (27 SNPs), and rs907091 was the only poly-miRTS located in the non-HLA region (17 chromosomes). We finally selected the pair of rs907091 and IKZF3 for functional validation due to the following considerations: the physical location of rs907091 in the 3’UTR of IKZF3, the top significant cis eQTL in LCLs (eQTL effect value: 62.12), the significant GWAS association (P=3.9E-08), their known functions of IKZF3 associated with autoimmune diseases (e.g., systemic lupus erythematosus, SLE)[33,34], and the corresponding miRNAs (miR-326 and miR-330-5p) were simultaneously predicted by TargetScan, miRanda, PITA and miRTarBase.
Table 1. The Results of eQTL, GWAS and Target miRNA for the 32 Identified Pairs Between 32 Unique Poly-miRTSs and 5 Target Genes
SNP.ID |
Chr |
Position |
Allele1 |
Allele2 |
OR.A1 |
GWAS P value |
eQTL target gene |
eQTL effect value |
eQTL type |
eQTL cell type |
Target miRNA |
rs907091 |
chr17 |
37921742 |
T |
C |
0.92 |
3.90E-08 |
IKZF3 |
62.12 |
cis |
LCLs |
hsa-miR-1266/211-3p/3144-5p/3191-5p/ 326/330-5p/4314/4497/4518 |
rs1059288 |
chr6 |
33267672 |
A |
G |
0.85 |
2.70E-25 |
TAPBP |
5.58 |
cis |
LCLs |
hsa-miR-1324/1972/2114-5p/3138/ 4520a-3p/4746-5p |
rs241453 |
chr6 |
32796226 |
A |
G |
0.9 |
1.00E-08 |
TAP2 |
6.48/6.14/5.57 |
cis |
Fibroblasts/T cells/LCLs |
hsa-miR-1302/4298 |
rs3091282 |
chr6 |
33057198 |
G |
C |
1.41 |
1.40E-85 |
HLA-DPB1 |
63.63 |
cis |
LCLs |
hsa-miR-3074-5p/516a-3p/516b-3p |
rs3091283 |
chr6 |
33057213 |
T |
C |
0.75 |
1.50E-55 |
HLA-DPB1 |
43.03 |
cis |
LCLs |
hsa-miR-1200/1272/3161/3682-3p/4468/617 |
rs3117228 |
chr6 |
33056435 |
T |
G |
0.71 |
1.60E-85 |
HLA-DPB1 |
63.6 |
cis |
LCLs |
hsa-miR-16-2-3p/195-3p/570-3p |
rs3117229 |
chr6 |
33056069 |
A |
G |
0.74 |
6.20E-57 |
HLA-DPB1 |
45.65 |
cis |
Monocytes |
hsa-miR-1243/301a-5p/4522/4714-5p |
rs3128964 |
chr6 |
33055818 |
A |
G |
1.41 |
1.60E-85 |
HLA-DPB1 |
63.64 |
cis |
LCLs |
hsa-miR-3667-3p/4297/5581-5p |
rs3128965 |
chr6 |
33055899 |
A |
G |
0.84 |
5.20E-16 |
HLA-DPB1 |
16.74 |
cis |
Monocytes |
hsa-miR-4775/590-3p |
rs3128966 |
chr6 |
33055946 |
A |
G |
0.84 |
5.20E-16 |
HLA-DPB1 |
16.66 |
cis |
Monocytes |
hsa-miR-26a-1-3p/26a-2-3p/617 |
rs3177928 |
chr6 |
32412435 |
A |
G |
1.25 |
3.00E-28 |
HLA-DRA |
12.43 |
cis |
Monocytes |
hsa-miR-21-3p/3591-3p/4768-5p/494/942 |
rs7195 |
chr6 |
32412539 |
A |
G |
0.53 |
1.00E-250 |
HLA-DRA |
91.53/39.31 |
cis |
Monocytes/LCLs |
hsa-miR-3652/4446-3p/4492/4728-3p/5001-5p/875-3p |
rs9277533 |
chr6 |
33054721 |
T |
C |
0.71 |
1.20E-85 |
HLA-DPB1 |
63.65 |
cis |
LCLs |
hsa-miR-194-5p/568 |
rs9277534 |
chr6 |
33054807 |
A |
G |
1.41 |
1.20E-85 |
HLA-DPB1 |
63.43 |
cis |
LCLs |
hsa-miR-3545-5p/4321/4460 |
rs9277537 |
chr6 |
33055009 |
A |
C |
0.71 |
1.30E-85 |
HLA-DPB1 |
63.67 |
cis |
LCLs |
hsa-miR-4477b/496/5002-5p |
rs9277538 |
chr6 |
33055047 |
A |
G |
1.4 |
4.60E-78 |
HLA-DPB1 |
63.77 |
cis |
LCLs |
hsa-miR-1243/4757-3p/934 |
rs9277539 |
chr6 |
33055079 |
A |
G |
1.31 |
2.00E-49 |
HLA-DPB1 |
43.16 |
cis |
LCLs |
hsa-miR-138-5p/3165/3691-5p/4456 |
rs9277540 |
chr6 |
33055123 |
A |
G |
1.41 |
1.30E-85 |
HLA-DPB1 |
56.08 |
cis |
LCLs |
hsa-miR-194-3p/3150a-3p/491-5p |
rs9277541 |
chr6 |
33055158 |
A |
G |
1.41 |
1.30E-85 |
HLA-DPB1 |
63.61 |
cis |
LCLs |
hsa-miR-199a-5p/221-3p/222-3p/4666a-5p |
rs9277546 |
chr6 |
33055346 |
T |
G |
1.41 |
1.30E-85 |
HLA-DPB1 |
63.73 |
cis |
LCLs |
hsa-miR-1243/361-5p/568 |
rs9277547 |
chr6 |
33055367 |
A |
C |
0.71 |
1.30E-85 |
HLA-DPB1 |
63.74 |
cis |
LCLs |
hsa-miR-3653/4270/4499/873-3p |
rs9277549 |
chr6 |
33055419 |
A |
G |
1.41 |
1.30E-85 |
HLA-DPB1 |
63.65 |
cis |
LCLs |
hsa-miR-552/764 |
rs9277550 |
chr6 |
33055487 |
T |
C |
1.44 |
4.60E-90 |
HLA-DPB1 |
141.66/63.38 |
cis |
Monocytes/LCLs |
hsa-miR-1185-5p/3679-5p/4534/4746-5p/4802-5p |
rs9277551 |
chr6 |
33055494 |
T |
G |
0.7 |
6.70E-90 |
HLA-DPB1 |
64.02 |
cis |
LCLs |
hsa-miR-2355-5p/4746-5p |
rs9277553 |
chr6 |
33055516 |
T |
C |
1.41 |
1.70E-85 |
HLA-DPB1 |
59.44 |
cis |
LCLs |
hsa-miR-4758-3p |
rs9277554 |
chr6 |
33055538 |
T |
C |
0.71 |
1.70E-85 |
HLA-DPB1 |
63.63 |
cis |
LCLs |
hsa-miR-3691-3p/4724-3p/5190/550b-2-5p |
rs9277555 |
chr6 |
33055605 |
A |
G |
0.75 |
2.40E-55 |
HLA-DPB1 |
43.1 |
cis |
LCLs |
hsa-miR-345-5p/4264/455-3p/670 |
rs9277566 |
chr6 |
33056916 |
T |
G |
0.71 |
1.50E-85 |
HLA-DPB1 |
63.57 |
cis |
LCLs |
hsa-miR-1276/140-5p/335-5p/548as-3p |
rs9296075 |
chr6 |
33056788 |
A |
G |
0.7 |
1.30E-21 |
HLA-DPB1 |
30.03 |
cis |
LCLs |
hsa-miR-4780 |
rs9296076 |
chr6 |
33056840 |
T |
C |
1.57 |
1.50E-50 |
HLA-DPB1 |
41.82 |
cis |
LCLs |
hsa-miR-1273e/1304-3p |
rs931 |
chr6 |
33054550 |
A |
G |
0.71 |
1.20E-85 |
HLA-DPB1 |
52.2 |
cis |
LCLs |
hsa-miR-132-3p/212-3p/5688/570-3p |
rs9501259 |
chr6 |
33055551 |
A |
G |
0.64 |
1.60E-50 |
HLA-DPB1 |
34.75 |
cis |
LCLs |
hsa-miR-5190 |
Notes: Based on the identified 114 pairs, we further identified 32 pairs between 32 unique poly-miRTSs and 5 target genes according to the positions of the SNP in the 3’UTR of their eQTL genes.
Table 2 shows the results of the allelic expression imbalance experiment. For each biological sample, repeated verification experiments were applied. At the DNA level, the mean allelic ratio of C/T was 0.83, and the mean ratios of NO.1 and NO.2 cDNA were 0.99 and 0.96, respectively. However, after normalization to the ratio in genomic DNA quantization, the allelic expression ratios of C/T in IKZF3 were 1.20 and 1.16 from the NO.1 and NO.2 cDNA samples, respectively. In conclusion, the C allele had a higher expression of ~18% than the T allele, suggesting an allelic expression imbalance.
Table 2. The Results of Allelic Expression Imbalance Experiment
Site |
Sample |
Allele 1 |
Allele 2 |
Height 1 |
Height 2 |
HC/HT |
Mean of DNA_HC/HT |
Correct HC/HT |
Mean of HC/HT |
|
rs907091 |
DNA |
Repeat 1 |
C |
T |
24267 |
28833 |
0.84 |
0.83 |
|
|
Repeat 2 |
C |
T |
24700 |
30186 |
0.82 |
|
|
|||
NO.1 cDNA |
Repeat 1 |
C |
T |
16849 |
16786 |
1.00 |
|
1.21 |
1.20 |
|
Repeat 2 |
C |
T |
22827 |
23204 |
0.98 |
|
1.19 |
|||
NO.2 cDNA |
Repeat 1 |
C |
T |
17231 |
18000 |
0.96 |
|
1.15 |
1.16 |
|
Repeat 2 |
C |
T |
24944 |
25934 |
0.96 |
|
1.16 |
|||
Notes: All the DNA and cDNA were from one rs907091 heterozygosis volunteer. NO.1 cDNA and No.2 cDNA represent two times of cDNA amplifications. Two repeats of AEI were performed for each cDNA. Height 1, electrophoretic peak height of Allele C; Height 2, electrophoretic peak height of Allele T; HC/HT, height value of Allele C/height value of Allele T; Correct HC/HT, after correcting the DNA level, height value of Allele C/height value of Allele T in cDNA level.
Furthermore, dual-luciferase reporter gene assays were performed to validate the putative regulatory mechanism between IKZF3 targeting the SNP rs907091 and the two predicted miRNAs (miR-326 and miR-330-5p). We constructed two dual-luciferase reporter plasmids containing the rs907091 T and C alleles. As shown in Figure 2, in the control group (no involved miRNA), no significant difference was observed between T and C alleles, but after treatment with miR-326 or miR-330-5p, the luciferase activity was significantly higher in the C allelic plasmid than in the T allelic plasmid (P<0.05). As shown in Figure 3, we proposed a biological hypothesis for the relationship among miR-326, miR-330-5p, rs907091 and IKZF3. The miR-326 and/or miR-330-5p can bind to the 3’UTR of the IKZF3 gene. However, the genotypes of rs907091 at the 3’UTR of the IKZF3 gene could influence the affinity between miRNA and target mRNA. The results suggested that with the regulatory role of miR-326 and/or miR-330-5p, rs907091 C>T in the 3’UTR could upregulate IKZF3 expression.
|
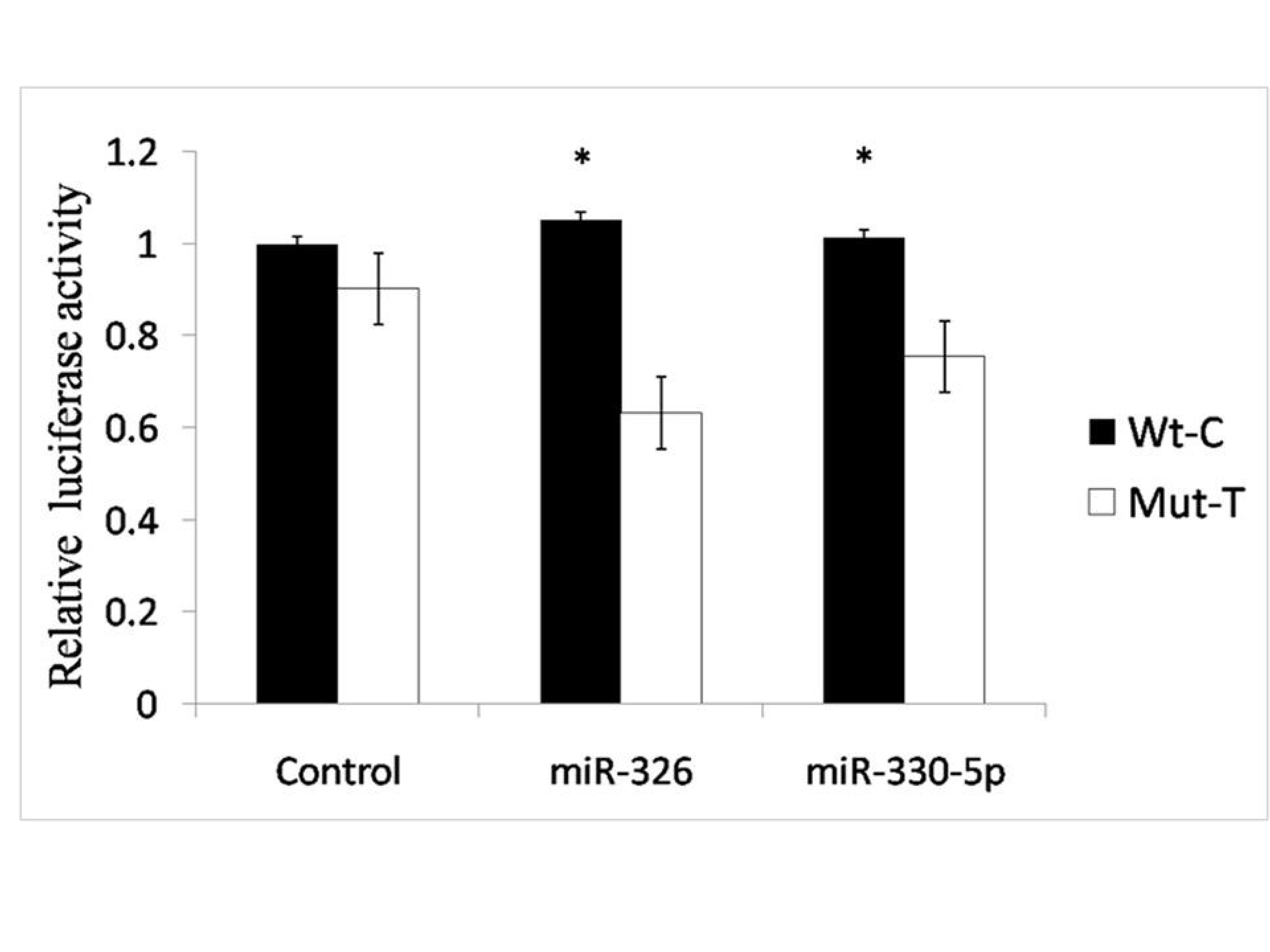
Figure 2. Dual-luciferase reporter assays for rs907091 in the 3’UTR of IKZF3. Wt-C, wild-type allele C; Mut-T, mutant allele T; Control, without the treatment of miRNA. Data were fold changes when compared with control Wt-C. All experiments were repeated three times, and each experiment had five biological replicates for each allele. * represents a significant difference between Wt-C and Mut-T (P<0.05). The bars represent SE.
|
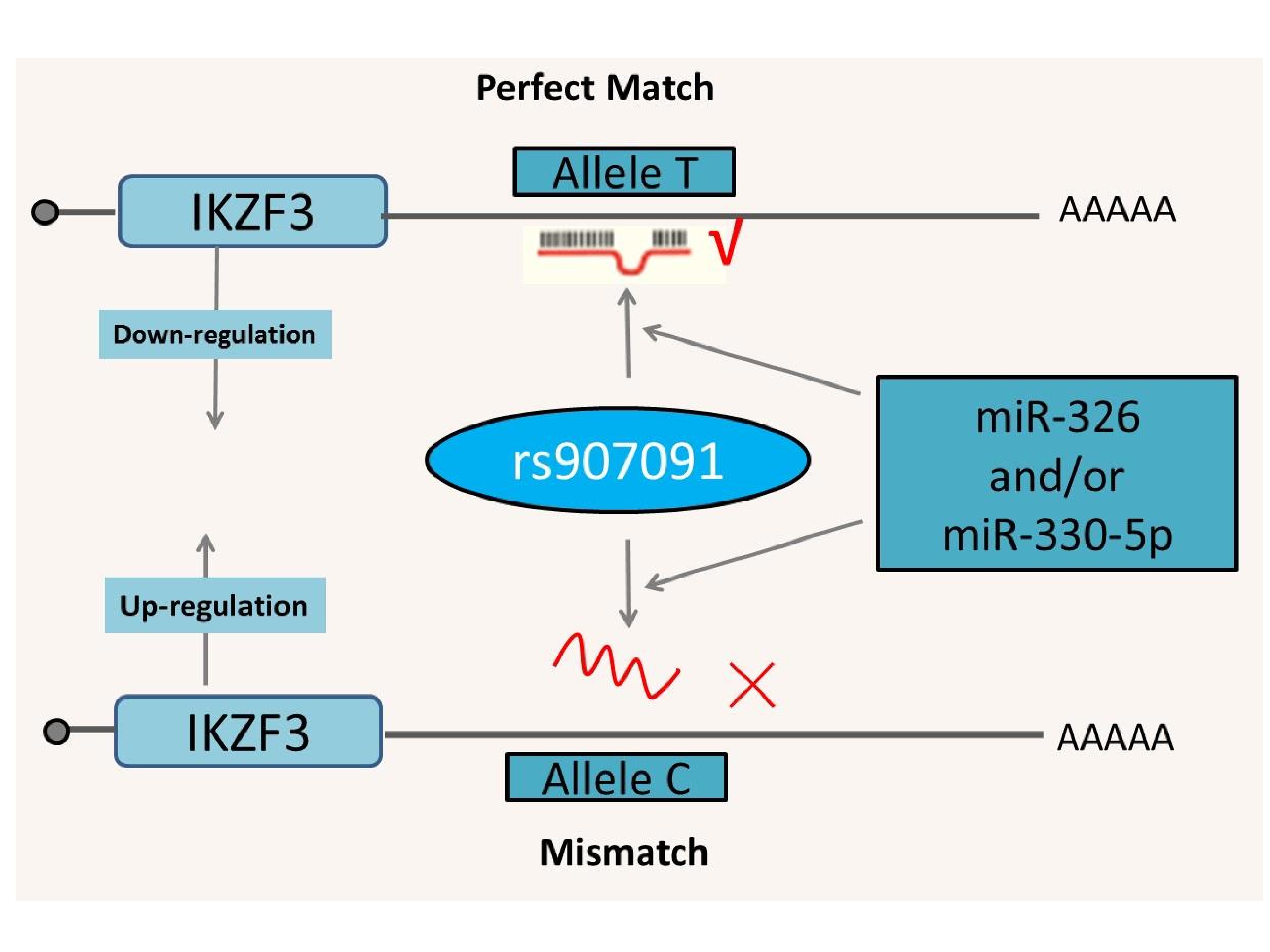
Figure 3. The proposed hypothesis for the relationship among miR-326 and miR-330-5p, rs907091 and IKZF3. miR-326 and/or miR-330-5p can bind to the 3’UTR of the IKZF3 gene. However, the genotypes of rs907091 at the 3’UTR of the IKZF3 gene could influence the affinity between miRNA and target mRNA and lead to allele-specific expression differences (rs907091 C>T upregulates IKZF3 expression).
4 DISCUSSION
Recently, GWAS has identified a large number of genetic loci associated with RA. However, exploring the functional mechanisms underlying the associations is a huge challenge. This study performed an integrative analysis and in-house molecular experiments to identify functional variants that are associated with the pathogenesis of RA through a miRNA-mediated regulatory mechanism.
Classic HLA loci are regarded as major contributors to the pathogenesis of immune diseases, including RA. However, recent studies have identified a series of candidate genes outside the HLA region, suggesting that non-HLA RA susceptibility loci may also be important and more specific in RA pathogenesis[35-37]. Therefore, we selected rs907091 in the non-HLA region for further in-house functional exploration. As we expected, AEI and dual-luciferase reporter gene assays, taken together, suggest that rs907091 is a potential functional variant for rheumatoid arthritis, which probably impacts its differential expression of target IKZF3 mediated by two miRNAs. We searched the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) database, and the results of GSE23561 indicated that IKZFR3 was differentially expressed between RA patients and healthy controls in peripheral blood (P<0.01)[38]. In addition, we downloaded the GSE91026 dataset (Global miRNA expression profiles of fibroblast-like synoviocytes in RA patients and osteoarthritis[39]). In this dataset, the expression level of miR-330-5p was significantly different between RA and OA (osteoarthritis) patients (P<0.05).
Previous GWASs have identified numerous genetic variants associated with RA, but most of the identified variants are located in noncoding regions, including in the 3’UTR. The combination of miRNAs and the 3’UTR of host genes is a common cis-acting mechanism in regulating gene expression. Therefore, polymorphisms in miRNA binding sites of 3’-UTRs could potentially influence the affinity between miRNAs and their target mRNAs[15,16]. This could also affect the efficiency of miRNAs regulating protein expression by altering their capacity to repress mRNA translation or promote mRNA decay[15,17]. Our dual-luciferase reporter gene assays validated that allelic differential interactions exist between rs907091 and IKZF3 expression. A similar result from a luciferase reporter assay revealed allele-specific regulation of IKZF3 hosting rs907091 mediated by miR-326[40]. Another study also observed the allelic imbalance of rs907091 in the expression level of IKZF3[41]. Taken together, these results suggest that the miRNA-mediated cis-acting mechanism may contribute to the RA association of rs907091 at IKZF3.
The IKZF3 gene encodes a member of the Ikaros family of zinc-finger proteins, transcription factors that are important in the regulation of B lymphocyte proliferation and differentiation[42]. IKZF3 deficiency mainly affects the function of B cells by increasing hyperactive immature B cell precursors and decreasing peritoneal, marginal and recirculating B cells[43-46]. It has been reported that Ikaros family members are essential for normal T cell development[47], and overexpression of Aiolos in Nalm-6 cells could inhibit cell proliferation and apoptosis and arrest the cell cycle at the G0/G1 phase[48]. It is well known that immune cells such as T and B lymphocytes are crucial in the pathogenesis and development of RA and are involved in the activation of immune cells and secretion of multiple cytokines. Furthermore, recent studies have shown that IKZF3 is associated not only with RA[49-51] but also with other autoimmune diseases[41,52-54].
In summary, we performed an integrative analysis and in-house molecular experiments to explore the functional mechanisms underlying the associations between poly-miRTSs and RA. We identified that the rs907091 variant in the 3’UTR of IKZF3 probably contributes to the pathogenesis of RA by binding to miR-326 and miR-330-5p. This study may provide helpful insights into the molecular functional genetic mechanisms underlying complex human diseases.
Acknowledgements
The study was supported by Natural Science Foundation of China (No.82373587, No.82173529, No.82173598, and No. 82103922).
Conflicts of Interest
The authors declared no conflict of interest.
Author Contribution
The authors made contributions to the conception and interpretation of the study and were involved in revising the manuscript. Lei S and Xia W designed the study and interpreted the data, and Xia W drafted the manuscript. Xia W and Lei S made a substantial contribution to the acquisition and analysis of the data. Lei S revised the manuscript critically for important intellectual content. All authors approved the final version of the manuscript.
Abbreviation List
3’-UTRs, 3’-Untranslated regions
AEI, Allelic expression imbalance experiment
eQTL, Expression quantitative trait locus analysis
gDNA, Genomic DNA
GWAS, Genome wide association study
LCLs, Lymphoblastoid cell lines
miRNAs, MicroRNAs
poly-miRTSs, Polymorphisms in miRNA target sites
PPI, Protein-protein interaction
RA, Rheumatoid arthritis
SNP, Single nucleotide polymorphisms
STRING, Search Tool for the Retrieval of Interacting Genes
References
[1] Eleftherohorinou H, Hoggart CJ, Wright VJ et al. Pathway-driven gene stability selection of two rheumatoid arthritis GWAS identifies and validates new susceptibility genes in receptor mediated signalling pathways. Hum Mol Genet, 2011; 20: 3494-3506.[DOI]
[2] Julià A, Ballina J, Cañete JD et al. Genome-wide association study of rheumatoid arthritis in the Spanish population: KLF12 as a risk locus for rheumatoid arthritis susceptibility. Arthritis Rheum-Us, 2008; 58: 2275-2286.[DOI]
[3] Gan RW, Demoruelle MK, Deane KD et al. Omega-3 fatty acids are associated with a lower prevalence of autoantibodies in shared epitope-positive subjects at risk for rheumatoid arthritis. Ann Rheum Dis, 2017; 76: 147-152.[DOI]
[4] López-Isac E, Martín J-E, Assassi S et al. Brief Report: IRF4 Newly Identified as a Common Susceptibility Locus for Systemic Sclerosis and Rheumatoid Arthritis in a Cross-Disease Meta-Analysis of Genome-Wide Association Studies. Arthritis Rheumatol, 2016; 68: 2338-2344.[DOI]
[5] Kochi Y, Okada Y, Suzuki et al. A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat Genet, 2010; 42: 515-519.[DOI]
[6] Gregersen P, Amos C, Lee A et al. REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet, 2009; 41: 820-823.[DOI]
[7] Saxena R, Plenge RM, Bjonnes AC et al. A Multinational Arab Genome-Wide Association Study Identifies New Genetic Associations for Rheumatoid Arthritis. Arthritis Rheumatol, 2017; 69: 976-985.[DOI]
[8] Xiao X, Hao J, Wen Y et al. Genome-wide association studies and gene expression profiles of rheumatoid arthritis: An analysis. Bone Joint Res, 2016; 5: 314-319 .[DOI]
[9] Okada Y, Terao C, Ikari K et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat Genet, 2012; 44: 511-516.[DOI]
[10] Stahl E, Raychaudhuri S, Remmers E et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet, 2010; 42: 508-514.[DOI]
[11] Zhernakova A, Stahl EA, Trynka G et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet, 2011; 7: e1002004.[DOI]
[12] Márquez A, Vidal-Bralo L, Rodríguez-Rodríguez L et al. A combined large-scale meta-analysis identifies COG6 as a novel shared risk locus for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis, 2017; 76: 286-294.[DOI]
[13] Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell, 2009; 136: 215-233.[DOI]
[14] Eulalio A, Huntzinger E, Nishihara T et al. Deadenylation is a widespread effect of miRNA regulation. RNA, 2009; 15: 21-32.[DOI]
[15] Richardson K, Nettleton JA, Rotllan N et al. Gain-of-function lipoprotein lipase variant rs13702 modulates lipid traits through disruption of a microRNA-410 seed site. Am J Hum Genet, 2013; 92: 5-14.[DOI]
[16] Miller CL, Haas U, Diaz R et al. Coronary heart disease-associated variation in TCF21 disrupts a miR-224 binding site and miRNA-mediated regulation. PLoS Genet, 2014; 10: e1004263.[DOI]
[17] Ghanbari M, de Vries PS, de Looper H et al. A genetic variant in the seed region of miR-4513 shows pleiotropic effects on lipid and glucose homeostasis, blood pressure, and coronary artery disease. Hum Mutat, 2014; 35: 1524-1531.[DOI]
[18] Brendle A, Lei H, Brandt A et al. Polymorphisms in predicted microRNA-binding sites in integrin genes and breast cancer: ITGB4 as prognostic marker. Carcinogenesis, 2008; 29: 1394-1399.[DOI]
[19] Yang YP, Ting WC, Chen LM et al. Polymorphisms in MicroRNA Binding Sites Predict Colorectal Cancer Survival. Int J Med Sci, 2017; 14: 53-57.[DOI]
[20] Manikandan M, Munirajan AK. Single nucleotide polymorphisms in microRNA binding sites of oncogenes: implications in cancer and pharmacogenomics. Omics, 2014; 18: 142-154.[DOI]
[21] Wang W, Li F, Mao Y et al. A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum Genet, 2013; 132: 641-648.[DOI]
[22] Xu T, Zhu Y, Wei QK et al. A functional polymorphism in the miR-146a gene is associated with the risk for hepatocellular carcinoma. Carcinogenesis, 2008; 29: 2126-2131.[DOI]
[23] Okada Y, Wu D, Trynka G et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature, 2014; 506: 376-381.[DOI]
[24] Li G, Diogo D, Wu D et al. Human genetics in rheumatoid arthritis guides a high-throughput drug screen of the CD40 signaling pathway. PLoS Genet, 2013; 9: e1003487.[DOI]
[25] Deng FY, Lei SF, Zhu H et al. Integrative analyses for functional mechanisms underlying associations for rheumatoid arthritis. J rheumatol, 2013; 40: 1063-1068.[DOI]
[26] Naranbhai V, Fairfax B, Makino S et al. Genomic modulators of gene expression in human neutrophils. Nat Commun, 2015; 6: 7545.[DOI]
[27] Liu C, Zhang F, Li T et al. MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPs and eQTLs. BMC Genomics, 2012; 13: 661.[DOI]
[28] Kirubamani NH, Meenatshi M. Target Scan-The Experience at Saveetha Medical College. J Clin of Diagn Res, 2013; 7: 873-875.[DOI]
[29] Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res, 2006; 34: W451-454.[DOI]
[30] O’Connor BD, Day A, Cain S et al. GMODWeb: a web framework for the Generic Model Organism Database. Genome Biol, 2008; 9: R102.[DOI]
[31] Huang D, Sherman B, Lempicki R. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc, 2009; 4: 44-57.[DOI]
[32] Szklarczyk D, Morris JH, Cook H et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res, 2017; 45: D362-D368.[DOI]
[33] Ye, L. X., Fu, C. W., Jiang, F. & Meng, W. Association between IKZF3 gene polymorphisms and systemic lupus erythematosus in Han ethnic group in southern China: a case-control study. Chinese J Epidemiol, 2016; 37: 996-1002.[DOI]
[34] Cai X, Qiao Y, Diao C et al. Association between polymorphisms of the IKZF3 gene and systemic lupus erythematosus in a Chinese Han population. PLoS One, 2014; 9: e108661.[DOI]
[35] Ahmadloo S, Taghizadeh M, Akhiani M et al. Single Nucleotide Polymorphism rs 2476601 of PTPN22 Gene and Susceptibility to Rheumatoid Arthritis in Iranian Population. Iran J Allergy Asthm, 2015; 437-442.
[36] Plant D, Flynn E, Mbarek H et al. Investigation of potential non-HLA rheumatoid arthritis susceptibility loci in a European cohort increases the evidence for nine markers. Ann Rheum Dis, 2010; 69: 1548-1553.[DOI]
[37] Snir O, Gomez-Cabrero D, Montes A et al. Non-HLA genes PTPN22, CDK6 and PADI4 are associated with specific autoantibodies in HLA-defined subgroups of rheumatoid arthritis. Arthritis Res Ther, 2014; 16: 414.[DOI]
[38] Grayson BL, Wang L, Aune T. Peripheral blood gene expression profiles in metabolic syndrome, coronary artery disease and type 2 diabetes. Genes Immun, 2011; 12: 341-351.[DOI]
[39] Hong BK, You S, Yoo SA et al. MicroRNA-143 and -145 modulate the phenotype of synovial fibroblasts in rheumatoid arthritis. Exp Mol Med, 2017; 49: e363.[DOI]
[40] Ghanbari M, Franco OH, De Looper HWJ et al. Genetic Variations in MicroRNA-Binding Sites Affect MicroRNA-Mediated Regulation of Several Genes Associated With Cardio-metabolic Phenotypes. Circ Cardiovasc Gene, 2015; 8: 473-486.[DOI]
[41] Keshari PK, Harbo HF, Myhr KM et al. Allelic imbalance of multiple sclerosis susceptibility genes IKZF3 and IQGAP1 in human peripheral blood. BMC Genet, 2016; 17: 59.[DOI]
[42] Cortes M, Wong E, Koipally J et al. Control of lymphocyte development by the Ikaros gene family. Curr Opin Immunol, 1999; 11: 167-171.[DOI]
[43] Wang JH, Avitahl N, Cariappa A et al. Aiolos regulates B cell activation and maturation to effector state. Immunity, 1998; 9: 543-553.[DOI]
[44] Sun J, Matthias G, Mihatsch MJ et al. Lack of the transcriptional coactivator OBF-1 prevents the development of systemic lupus erythematosus-like phenotypes in Aiolos mutant mice. J Immunol, 2003; 170: 1699-1706.[DOI]
[45] Cariappa A, Tang M, Parng C et al. The follicular versus marginal zone B lymphocyte cell fate decision is regulated by Aiolos, Btk, and CD21. Immunity, 2001; 14: 603-615.[DOI]
[46] Narvi E, Nera KP, Terho P et al. Aiolos controls gene conversion and cell death in DT40 B cells. Scand J Immunol, 2007; 65: 503-543.[DOI]
[47] Mitchell JL, Seng A, Yankee TM. Expression patterns of Ikaros family members during positive selection and lineage commitment of human thymocytes. Immunol, 2016; 149: 400-412.[DOI]
[48] Zhuang Y, Li D, Fu J et al. Overexpression of AIOLOS inhibits cell proliferation and suppresses apoptosis in Nalm-6 cells. Oncol Rep, 2014; 31: 1183-1190.[DOI]
[49] Olmos L, Sayed M, Lee R. Reply to ‘Comment on: Long-term outcomes of neovascular glaucoma treated with and without intravitreal bevacizumab’. Eye, 2016; 30: 897-898.[DOI]
[50] Kurreeman FAS, Stahl EA, Okada Y et al. Use of a multiethnic approach to identify rheumatoid- arthritis-susceptibility loci, 1p36 and 17q12. Am J Hum Genet, 2012; 90: 524-532.[DOI]
[51] Yao M, Huang X, Guo Y et al. Disentangling the common genetic architecture and causality of rheumatoid arthritis and systemic lupus erythematosus with COVID-19 outcomes: Genome-wide cross trait analysis and bidirectional Mendelian randomization study. J Med Virol, 2023; 95: e28570.[DOI]
[52] Cai X, Liu X, Du S et al. Overexpression of Aiolos in Peripheral Blood Mononuclear Cell Subsets from Patients with Systemic Lupus Erythematosus and Rheumatoid Arthritis. Biochem Genet, 2016; 54: 73-82.[DOI]
[53] Farh KH, Marson A, Zhu J et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature, 2015; 518: 337-343.[DOI]
[54] Wang X, Zhu L, Ying S et al. Increased RNA editing sites revealed as potential novel biomarkers for diagnosis in primary Sjögren's syndrome. J Autoimmun, 2023; 138: 103035.[DOI]
Copyright © 2024 The Author(s). This open-access article is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, sharing, adaptation, distribution, and reproduction in any medium, provided the original work is properly cited.


